
Macrophage origin, phenotypic diversity, and modulatory signaling pathways in the atherosclerotic plaque microenvironment

Abstract
Atherosclerosis is the main pathological basis of most cardiovascular diseases and the leading health threat in the world. Of note, lipid-lowering therapy cannot completely retard atherosclerosis progression, even in patients treated with combined statins and PCSK9 inhibitors. This failure further impels researchers to explore other underlying therapeutic strategies except for lipid-lowering. Monocytes and macrophages are the major immune cell groups in atherosclerotic plaques. They play important roles in all stages of atherosclerosis, including the occurrence, advance, and regression. It is interesting that macrophages are demonstrated to have plastic and heterogenous characteristics within the dynamic atherosclerotic plaque microenvironment. Furthermore, the phenotype of macrophages can switch upon different microenvironmental stimulus. Therefore, macrophages have become a potential therapeutic target for anti-atherosclerosis treatment. This article reviews the phenotypic diversity of macrophages and their roles in dynamic atherosclerotic plaque microenvironment, especially the related signaling pathways involved in macrophage polarization and compounds exhibiting therapeutic effects.
Keywords
INTRODUCTION
Atherosclerosis is a progressive inflammatory disease that mainly affects large- and medium-sized arteries[1-3]. This disease is characterized by the formation of atherosclerotic plaques, which are composed of lipids, necrotic nuclei, calcified areas, inflammatory cells, endothelial cells, immune cells, and foam cells[2,3]. Monocytes-derived macrophages are the most abundant type of immune cells in atherosclerotic plaques and play a key role in the progression and regression of atherosclerosis[4,5].
Atherosclerosis is initiated by deposition of cholesterol-rich lipoproteins, such as oxidized low-density lipoproteins (ox-LDL), in the arterial wall[6,7]. This pathological alteration promotes the production of cytokines and chemokines that can recruit monocytes from circulation. In the following, monocytes can differentiate into macrophages upon the stimuli of the local atherosclerotic plaque microenvironment[6,7]. Mature macrophages express a variety of scavenger receptors, such as scavenger receptor A (SR-A) and cluster of differentiation (CD) 36, which are responsible for scavenging modified lipoproteins within the plaque[1,8,9]. Accumulation of neutral lipids in macrophages promotes the formation of foam cells, which is considered as a protective mechanism for clearance of deposited lipids from the arterial wall[1,8,9]. However, excessive accumulation of foam cells in the arterial wall leads to the formation of advanced atherosclerotic plaques[1]. Of note, the total number of macrophages is associated with the progress and severity of the plaque, and symptomatic plaques have more macrophages than asymptomatic plaques[10-12]. These atherosclerotic plaques cause narrowing of the arteries, which limits or stops blood flow to the tissue, inducing various cardiovascular events[13].
Atherosclerotic plaque microenvironmental stimuli influence the phenotypes and functions of macrophages, which in turn play key roles in atherosclerotic plaque progression and regression[6,14,15]. Macrophages are simply divided into classically activated (M1) and alternatively activated (M2) subtypes, which have been extensively studied since the end of the last century[16-18]. In terms of numbers, the pro-inflammatory M1 macrophages are enriched in progressing plaques, while the anti-inflammatory M2 macrophages are dominant in regressing plaques[19-21]. Moreover, accumulating evidence suggests that the phenotypic shift of plaque macrophages from an M1 phenotype to an M2 phenotype appears to play important roles in maintaining plaque stability and promoting atherosclerosis regression[16-18]. Over the past decade, the original classification of macrophages (M1 and M2) has been challenged due to the gradual discovery and identification of other macrophage subpopulations. These subpopulations of macrophages possess distinctive functional phenotypes in response to specific signals of the atherosclerotic plaque microenvironment[2,3,21-23].
Presently, the therapeutic strategies for atherosclerosis are mainly aimed at lipid-lowering. However, clinical data demonstrate that these strategies are not powerful enough to inhibit the progression of atherosclerosis and especially to promote its regression[24,25]. As mentioned above, the macrophages with plasticity in the dynamic atherosclerotic plaque microenvironment may be a potential therapeutic target. In this review, we summarize the recent advances in macrophage heterogeneity, macrophage metabolism, macrophage polarization, and potential modulatory signaling pathways in the context of atherosclerosis.
MONOCYTES RECRUITMENT AND DIFFERENTIATION INTO MACROPHAGES
Monocytes are derived from hematopoietic stem cells in the bone marrow and play important roles in the defense system. Monocytes exude blood vessels and enter tissues and organs, where they further differentiate into macrophages and become the most phagocytic cells in the body[26]. Monocytes and macrophages are involved in the progression of cardiovascular disease, and they are associated with the development of atherosclerotic plaques[27].
In mice blood, monocytes, especially the classical Ly6C+ cells, are generated by bone marrow. The migration of these monocytes from bone marrow requires C-C motif chemokine receptor 2 (CCR2), which is expressed only in Ly6C+ cells[28]. Furthermore, mitogen-activated protein kinase kinase kinase 8 (MAP3K8) deletion affects the number of monocytes and the production of inflammatory cytokines in the atherosclerotic lesions via decreasing CCR2 mRNA and protein levels, suggesting the important role of MAP3K8 in this process[26]. In human blood, monocytes are generated by bone marrow and spleen, and they can be classified into at least three types: classical monocytes (CD14hiCD16-cells), intermediate monocytes (CD14++CD16+ monocytes), and non-classical monocytes (CD14lowCD16++ cells)[15,29,30]. Classical mononuclear cells account for the majority of these cells. Recently, multicolor flow cytometry and mass cytometry analysis have further improved our knowledge about monocyte subtypes[31,32]. For instance, Hamers et al.[33] recently described eight human monocyte subtypes distinguished by 34 unique surface markers. Furthermore, the expansion of Slan+CXCR6+ nonclassical monocytes in individuals is associated with coronary artery disease. Accumulating studies have demonstrated the phenotypic diversity of monocytes and highlights the unique migratory and efferocytotic capacity of each subtype, which may ultimately influence the development tendency of atherosclerosis[4,34,35].
In the case of atherosclerosis, when monocytes adsorb or invade the endothelium, the autoregulatory and anti-thrombotic effects of endothelial cells are weakened, thereby promoting the occurrence of atherosclerosis[4,36]. Mononuclear cells have obvious deformable characteristic and are responsible for clearing injured, senescent, dead cells, and cell debris. Upon atherosclerosis, the recruited monocytes are predominantly pro-inflammatory cells that have a high invasive capacity to enter tissues and become macrophages[37-39]. The recruitment of monocytes is critical to the formation of atherosclerosis plaques. It has been demonstrated that the number of circulating monocytes is positively related to the number of macrophages in the atherosclerotic plaques[40]. Multiple factors, such as hypercholesterolemia, hyperglycemia, and life habits (e.g., smoking), can influence monocyte recruitment.
Hypercholesterolemia is an important factor contributing to atherosclerosis[24,41]. Accumulation of lipids, especially cholesterol, in the arterial wall is the main feature of atherosclerosis. In response to lipid accumulation, monocytes and macrophages exhibit different gene expression and lipid metabolism[42]. For example, high lipoprotein X in hypercholesterolemia drives monocytes changes, such as upregulation of SR-A and CD36, which enhance uptake of modified low-density lipoprotein (LDL), such as ox-LDL, leading to the formation of foam cells[43-45]. Of note, ox-LDL induces monocyte activation through abnormal regulation of the dual specificity phosphatase-1 and p38 mitogen-activated protein kinase (MAPK) signaling axis[46]. Accumulation of monocytes under the endothelium, leading to plaque development, and the following activation of smooth muscle cells contribute the majority of foam cells in mice atherosclerotic plaques[47,48].
The production of reactive oxygen species (ROS) induced by obesity and hyperglycemia may contribute to the induction of M1-like pro-inflammatory macrophages by activating the related signaling pathways[49]. Significantly, diabetes mellitus is characterized by an increase in inflammatory monocytes/macrophages as well as high levels of tumor necrosis factor (TNF) α, interleukin (IL)-1β, and IL-6[50-52]. These alterations, such as activated Ly6Chigh monocytes, further contribute to hyperglycemia and insulin resistance[53,54]. Of note, high-density lipoprotein reduces proliferation of bone marrow progenitor cells via inhibiting cholesterol accumulation, and it decreases the production of monocytes and their general recruitment to the inflammatory sites, thus alleviating atherosclerosis[54].
Smoking influences all stages of atherosclerotic plaque formation[55,56]. Cigarette smoking is associated with vascular endothelial dysfunction, increased expression of monocyte tissue factor, and oxidative stress due to the increased ROS production and decreased antioxidant defense systems[57,58]. These changes have an important effect on monocyte recruitment. Cigarette smoke condensate can induce the differentiation of THP-1 monocytes into macrophages and significantly increase the levels of CD14 and CD36. In addition, smoking-induced expression of miR-124-3p in circulation can induce monocyte transformation into foam cells, contributing to the formation of atherosclerotic plaques[59]. Furthermore, cigarette smoke condensate may activate the NLRP3 inflammasome and production of high levels of IL-1β and IL-18 possibly via the myeloid differentiation primary response 88/nuclear factor kappa-B (NF-κB) pathway[56,60]. Notably, the activated NLRP3 inflammasome may enhance the levels of scavenger receptors, cholesterol esterification, and matrix metalloproteins, thereby promoting atherosclerosis progression[61].
MACROPHAGE PHENOTYPIC DIVERSITY AND FUNCTIONS
Accumulation of macrophages within the arterial wall is a characteristic feature of atherosclerosis. Previously, macrophages are classified into two main types, designated as pro-inflammatory M1 and anti-inflammatory M2 macrophages[6,62-64]. In fact, atherosclerosis macrophages are influenced by multiple plaque microenvironmental stimuli, such as oxidized lipids, cytokines, and senescent erythrocytes[21-23]. Growing evidence demonstrates that atherosclerotic plaque microenvironment can induce distinct subpopulations of macrophages, as summarized in Table 1.
Macrophage phenotype, markers, secreta, and functions
| Phenotype | Stimuli | Human markers | Mice markers | Cytokines, chemokines, and other secreted mediators | Functions |
| M1 | IFN-γ, LPS, GM-CSF, TNF-α | IL1β, TNF, IL6, IL12, IL23, CXCL9, CXCL10, CXCL11, SOCS1, MARCO, TLRs, iNOS | IL1β, TNF, IL6, IL12, IL23, CXCL9, CXCL10, CXCL11, Arg II, TLRs, iNOS | TNF-α, IL (1β, 6, 12, 23), CCL (2, 3, 4, 5, 8, 9, 10, 11), MMP (1, 3, 9), iNOS, ROS | Pro-inflammation, host defense, tumor resistance, bacterial killing |
| M2a | IL-4, IL-13 | CD68, CD163, MHC II, MR, CD200R, IL-1Ra, CCL18, Dectin-1, IL-1R II, DC-SIGN | Arg-1, resistin-like α, Ym1, Ym2, MMGL, stabilin1, CD163, Dectin-1, Chil3, FIZZ1 | IL10, TGF-β, IL-1Ra, CCL17, CCL18, CCL22, CCL24 | Anti-inflammatory, tissue repair, wound healing, allergy, parasite killing |
| M2b | Immune complexes, TLR Ligands, IL-1β | CCL1, IL-10 high/IL-12 low, TNF-α, CD86, IL-6, MHC II, MR | CCL1, IL-10 high/IL-12 low, TNF-α, CD86, IL-6, MHC II, MR, LIGHT | TNF-α, IL-1β, IL-6, IL-10, CCL1 | Immunoregulation, phagocytosis, iron-storage, high oxidative capactiy |
| M2c | IL-10, TGF-β, glucocorticoids | CXCL13, MR, CD163, IL-10, TGF-β, MerTK, TLRs, Tie-2 | CXCL13, MR, CD163, IL-10, TGF-β, MerTK, Arg-1 | IL-10, TGF-β, CCL16, CCL18, CXCL13,PTX3 | Immunoregulation, phagocytosis, matrix deposition, tissue remodeling, efferocytosis capacity |
| M2d | LPS+A2R ligands, IL-6 | VEGF, IL-10, TGF-β | VEGF, IL-10, TGF-β, iNOS | IL-10, VEGF, iNOS | Immunosuppression, phagocytosis, tissue remodeling, Protumoral, proangiogenic capacity |
| M4 | CXCL4 | MR, MMP7, S100A8 | MR, MMP7, S100A8 | MMP12, IL-6, TNF-α, MMP-7 | Proinflammatory, cytotoxicity, monocyte recruitment |
| Mox | Oxidized phospholipids | HMOX-1, Nrf2, Srxn1, Txnrd1 | HMOX-1, Nrf2, Srxn1, Txnrd1 | IL-10, IL-1β, COX-2 | Proatherogenic, proinflammatory, antioxidant |
| Mhem | Heme | CD163, ATF-1, MCH II | CD163, ATF-1, MCH II | IL-10; HMOX-1, LXRβ | Antioxidant erythrophagocytosis, atheroprotective |
| M(Hb) | Hemoglobin/haptoglobin complexes | CD163, MR | CD163, MR | IL-10, HMOX-1, LXRα, ABCA1, ABCG1 | Haemoglobin clearance, antioxidant, atheroprotective |
Classically activated M1 macrophages are mainly induced by microbial products such as cytokines TNF and interferon (IFN) γ or lipopolysaccharide (LPS)[63,64]. These macrophages produce high levels of IL-12 and IL-23, low levels of IL-10, and secrete the pro-inflammatory cytokines TNF, IL-6, and IL-1β[22,65]. Functionally, M1 macrophages are responsible for the removal of pathogens during infection by activating the NADPH oxidase system and subsequent production of ROS[66]. However, these reactive oxygen and nitrogen species generated by M1 macrophages may worsen oxidative stress in the atherosclerotic plaques[23]. With the development of inflammation, IL-10, IL-4, or IL-13 alternatively activates M2 phenotype macrophages that bearing anti-inflammatory functions. These cells express IL-10, transforming growth factor (TGF) β and arginase 1 (ARG1)[21]. M2 macrophages can be further divided into M2a, M2b, M2c, and M2d subtypes; each subtype is induced by different cytokines[67]. Of note, M2a, M2b, and M2c subtypes play key roles in anti-inflammation and immune regulation[68]. M2a macrophages are activated by IL-4 and IL-13, and they produce IL-10 as well as low levels of IL-12. These cells express high levels of mannose receptor (CD206), fibrotic factors, and TGF-β, which participate in tissue repair and wound healing[69-72]. Furthermore, these cells display high phagocytic capacity and low cholesterol efflux capacity due to the decreased expression of ATP-binding cassette (ABC) transporters. Immune complexes or toll-like receptor (TLR) activation in combination with IL-1β or LPS induces M2b macrophages, which express high levels of anti-inflammatory cytokine IL-10[73]. M2c macrophages are activated by glucocorticoid, IL-10, or TGF-β and produce anti-inflammatory factors[74,75]. M2b and M2c macrophages express high levels of proto-oncogene tyrosine protein kinase MER, a major apoptotic cell receptor, contributing to their phagocytic and immunomodulatory capacity[15,21,26,65,76]. M2d macrophages are stimulated by IL-6 and adenosine A(2A) receptor activation (e.g., agonists), characterized by the increased expression of IL-10 and vascular endothelial growth factor and reduced expression of TNF and IL-12[77,78]. These cells play a key role in angiogenesis[78,79]. Additionally, macrophage subpopulations have also been classified based on hemostatic roles of macrophages, including host defense macrophages (M1), wound healing macrophages (M2a), and immune regulatory macrophages (M2b/c)[73]. However, recent studies demonstrated that M2 macrophages are not always protective[80].
In recent years, oxidized phospholipids have been reported to induce Mox macrophages via the transcription factor nuclear erythroid-2 related factor[81]. Mox macrophages exhibit a morphological structure and biological functions that differ from M1 and M2 macrophages. In hypercholesterolemia mice, Mox macrophages account for about 30% of the total macrophages. These cells express pro-inflammatory markers, such as IL-1β and cyclooxygenase 2, and exhibit defective phagocytosis and chemotaxis[81]. Rupture of microvessels within the atherosclerotic plaque releases erythrocytes, which can be phagocytosed by macrophages, and then induce these cells into M(Hb) and Mhem phenotypes[82,83]. In vitro, stimulation of human monocytes by the hemoglobin-haptoglobin complex induces M(Hb) phenotype macrophages, which produce anti-inflammatory factors, such as cytokine IL-10, by activating the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT/PKB) signaling pathway[82,84-86]. These cells have increased activity of liver X receptor (LXR) α and ABC transporters, which result in increased cholesterol efflux and reduced lipid accumulation. Furthermore, they have increased ferroportin expression, which reduces intracellular iron accumulation upon iron exposure[82,87]. During hemorrhage, erythrocytes are released and scavenged by macrophages, causing an elevation in iron content and release of heme, which drive macrophages towards a Mhem phenotype[88,89]. Mhem cells are generally resistant to be transformed into foam cells due to the low oxidative stress inside the cells, and they are supposed to be atheroprotective macrophages[83]. M4 macrophages are activated by the chemokine C-X-C motif ligand (CXCL) 4, and they are characterized by expression of MMP-7 and the calcium binding protein S100A8. These cells display pro-inflammatory and pro-atherogenic properties[90].
The distribution of macrophage subtypes in the atherosclerotic lesion is not uniform. In human atherosclerotic plaques, M1 macrophages are located at the rupture-prone shoulders of mature plaques, while the M2 macrophages are away from the lipid core[10,91]. Macrophages in the fibrous cap surrounding the necrotic core seem to express equal numbers of M1 and M2 markers[10]. Accumulating studies suggested that pro-inflammatory M1 macrophages predominate in progressing atherosclerosis, whereas atheroprotective M2 macrophages are dominant in regressing plaques[11,92,93]. Functionally, M1 macrophages induce plaque inflammation, while M2 macrophages scavenge apoptotic cells and cell debris, improving tissue repair and healing[70,75,94]. Therefore, plaque M2 macrophages contribute to not only the maintenance of efficient efferocytosis but also the resolution of inflammation[95,96]. For instance, in the hemorrhagic areas of atherosclerotic plaques, ingestion of iron by M1 macrophages results in an uncontrolled pro-inflammatory phenotype and impairs wound healing, while M2 macrophages phagocytose both apoptotic cells and senescent erythrocytes via elevating ferroportin expression[94,97,98].
In advanced plaques, macrophages promote elastic fiber degradation and thinning of the fiber cap by increasing the production of matrix metalloproteinases[99]. These products cause macrophage invasion and tissue destruction, thereby contributing to the progression and even rupture of atherosclerosis[99,100]. Furthermore, M1 macrophages promote the initial calcium deposition in the necrotic core, named as microcalcification, whereas M2 macrophages promote the formation of macrocalcification by inducing osteoblast differentiation and mature of vascular smooth muscle cells. Macrocalcification induced by M2 macrophage leads to stable plaque, while microcalcification induced by M1 macrophages may lead to plaque rupture[101-103]. It seems that the evolution and regression/stabilization of plaques is associated in general with the prevalence of M1 and M2 macrophages, respectively. However, the biology of atherosclerotic plaques is very complex and involves myriad cellular, soluble, and functional factors that are still far from being completely deciphered. Given the fact that M2 phenotype macrophages are not always protective[80], the biological mechanisms underlying these phenomena are much more complex and not exclusively linked to macrophage phenotypes.
MACROPHAGE METABOLISM AND POLARIZATION
Metabolism is a network of biochemical reactions required to generate energy and other metabolic products. Despite the plasticity of the metabolic network, macrophages generally prefer particular metabolic pathways to match their functions[104]. In recent years, accumulating data suggest different metabolic microenvironments may finally regulate macrophage polarization through signaling cascades and epigenetic reprogramming. The metabolic characteristics of M1 and M2 macrophages are shown in Figures 1 and 2, respectively.
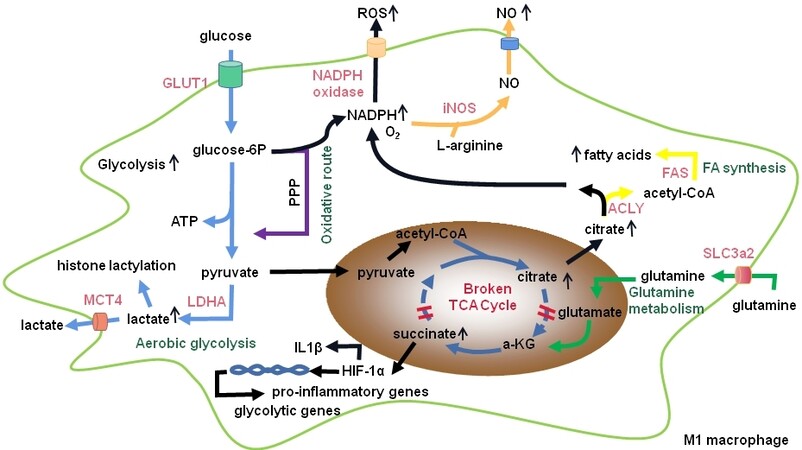
Figure 1. M1 macrophage metabolism. M1 macrophage metabolism is characterized by enhanced glycolysis and increased flux through the pentose phosphate pathway (PPP) to produce NADPH, which is used to produce the inflammatory mediators nitric oxide (NO) and reactive oxygen species (ROS). In M1 macrophages, the tricarboxylic acid (TCA) cycle is truncated, which leads to the accumulation of succinate and citrate metabolites. Succinate accumulation leads to stabilization of hypoxia-inducible factor 1 alpha (HIF-1α) and transcription of pro-inflammatory and glycolytic genes, whereas citrate is used for synthesis of fatty acids. ACLY: ATP-citrate lyase; α-KG: α-ketoglutarate; FAS: fatty acid synthase; GLUT: glucose transporter; iNOS: inducible nitric oxide synthase; LDH: lactate dehydrogenase; MCT: monocarboxylate transporter; SLC: solute carrier.
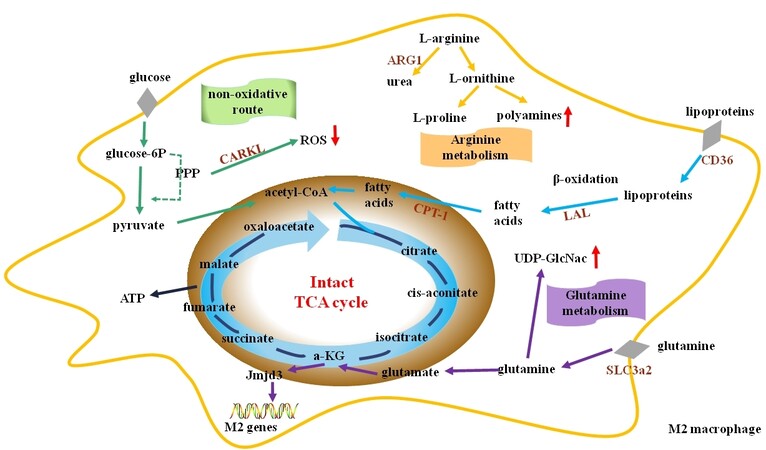
Figure 2. M2 macrophage metabolism. M2 macrophages rely on fatty acid oxidation (or β-oxidation) and L-glutamine metabolism to fuel the TCA cycle, which provide ATP for the cells. The pentose phosphate pathway (PPP) of M2 macrophages is decreased. Arginase 1 (ARG1) is highly expressed in M2 macrophages and competes with iNOS for their substrate L-arginine, thereby reducing NO production and leading to the production of urea and L-ornithine. α-KG: α-ketoglutarate; ATP: adenosine triphosphate; CARKL: carbohydrate kinase-like protein; CPT: carnitine palmitoyl transferase; LAL: lysosomal acid lipase; ROS: reactive oxygen species; SLC3a2: solute carrier family 3 member 2; TCA: tri-carboxylic acid; UDP-GlcNAc: uridine diphosphate N-acetylglucosamine.
Glycolysis is termed as the anaerobic breakdown of glucose into two molecules of pyruvate and ATP. In the following, pyruvate is converted into lactate under anaerobic conditions (also known as Warburg effect), and it is converted into acetyl coenzyme A and enters tri-carboxylic acid (TCA) cycle under aerobic conditions[105]. Glycolysis provides ATP at a faster pace compared with TCA cycle. Macrophages are essentially glycolytic cells in terms of bioenergetics. M1 macrophages display a metabolic shift towards the anaerobic glycolytic pathway to meet their rapid energy requirements [Figure 1]. In atherosclerotic plaques, hypoxia-activated hypoxia-inducible factor 1 alpha (HIF-1α), driven through NF-κB, is a key regulator of the glycolytic gene program. For instance, HIF-1α induces glucose transporter 1 (GLUT1), which is a rate-limiting glucose transporter and is upregulated in hypoxia-induced inflammatory macrophages to increase glucose levels[106]. Of note, glycolysis routes the carbon flux into the oxidative pentose phosphate pathway (PPP), which induces the generation of ROS via NADPH oxidases. M1 macrophages are characterized by enhanced PPP due to the high glycolytic flux. Carbohydrate kinase-like protein (CARKL) stimulates PPP to its non-oxidative route, thereby reducing ROS involved in pro-inflammatory activation and contributing to the macrophage polarization[107,108]. In M1 macrophages, the expression of CARKL is reduced, and overexpression of CARKL may promote macrophages to an M2 phenotype in line with the reduction of pro-inflammatory cytokines[109]. Furthermore, NADPH generated by PPP is a cofactor for inducible nitric oxide synthase (iNOS) to catabolize arginine and can be used to produce the antioxidant glutathione that plays a key role in maintaining redox homeostasis[104]. Therefore, NADPH can be used for nitric oxide (NO) and ROS production and may also support the redox balance in M1 macrophages. Except for energy source, lactate, the metabolic by-product of glycolysis, plays a key role in histone lactylation[110]. Compared to M1 macrophages, M2 macrophages display a decreased flux through the PPP and lower glycolytic rates. Furthermore, glycolytic stimulation may be not a requirement for M2 macrophage differentiation[111].
M2 macrophages sustain ATP production through the TCA cycle due to slower rates of aerobic glycolysis. It is suggested that M1 macrophages mainly depend on the TCA cycle for ROS generation, while M2 macrophages mainly relies on the TCA cycle for energy[105]. In M1 macrophages, TCA cycle is truncated, which leads to the accumulation of succinate and citrate metabolites. Citrate in M1 macrophages promotes the synthesis of the characteristic metabolite itaconic acid, which is an inducer for intracellular succinate accumulation[104]. Of note, the accumulated succinate can stabilize HIF-1α via induction of ROS and increased glycolytic flux, as reviewed by Geeraerts et al.[104]. In macrophages, HIF-1α regulates the expression of iNOS, which acts on L-arginine and promotes the production of the inflammatory factor NO and L-citrulline, contributing to the macrophage polarization towards the pro-inflammatory M1 phenotype. Interestingly, the product NO orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase[112]. Furthermore, LPS-induced succinate stabilizes HIF-1α and contributes to IL-1β production during inflammation[113]. On the contrary, HIF2α is suggested to be associated with an M2 phenotype macrophages[109]. Of note, HIF2α can regulate the transcription of ARG1, which is highly expressed in M2 macrophages and completes with iNOS for their substrate L-arginine, thereby reducing NO production and leading to the production of urea and L-ornithine. L-ornithine is a precursor of L-proline, which can be used for collagen synthesis, thereby contributing to wound healing[114]. Interestingly, the TCA cycle undergoes a two-stage remodeling in response to LPS and INF-γ stimulation; this metabolic reprogramming provides insights into how changing metabolism can regulate the functional transitions in macrophages[115].
The nonoxidative PPP provides intermediates used for nucleotide and amino acid synthesis[104]. Citrate can be exported from the mitochondria and used for fatty acid synthesis through several enzymes, as recently reviewed by Ménégaut et al.[116]. For example, citrate-derived acetyl-CoA can regulate the activation of chromatin-modifying enzymes and the biosynthesis of fatty acids. Of note, M1 macrophages are associated with the activation of fatty acid synthesis due to the increased substrate availability and upregulated genes that encode fatty acid synthesis. Due to the important role of citrate, M1 macrophages develop mechanisms to maintain the high intracellular citrate levels. However, fatty acid synthesis mediated by fatty acid synthase may induce the expression of pyrin domain containing 3 (NLRP3) and secretion of IL-1β, contributing to increased inflammation. For instance, glycine made from serine can induce glutathione synthesis, which results in IL-1β production[117]. Excessive HIF-1α-mediated fatty acid and triglyceride synthesis may also induce neutral lipid storage and foam cell formation[116]. Presently, most studies suggest that fatty acid synthesis in macrophages may be proatherogenic. The effects of amino acids on macrophage polarization were recently reviewed by Kieler et al.[118].
Mitochondrial oxidative phosphorylation is the primary mode of cellular energy production. However, this process is blocked in activated M1 macrophages, rendering them unable to be converted into M2 phenotype[106]. Mitophagy alters mitochondrial metabolism and occurs during M1 macrophage polarization. Inhibition of mitophagy increases mitochondrial mass and decreases the expression of glycolysis genes and inflammatory responses[119]. M2 macrophages have more mitochondria and increased oxygen consumption rates when compared to M1 macrophages. These differences provide M2 macrophages with great plasticity[105]. Blocking oxidative metabolism not only blocks the M2 phenotype but also drives the macrophage into an M1 phenotype[109]. It is suggested that microRNA-generating enzyme Dicer prevents atherosclerosis via enhancing fatty acid-fueled mitochondrial respiration in M2 macrophages of the apolipoprotein E-deficient mice[120]. Using real-time extracellular flux analysis, M1 macrophages are demonstrated to have enhanced glycolytic and decreased mitochondrial activity, whereas M2 macrophages display high mitochondrial oxidative phosphorylation[121].
Fatty acid oxidation (also known as β-oxidation) and glutamine metabolism provide important energy sources for macrophage polarization towards an M2 phenotype. During this process, triacylglycerol-rich lipoproteins, such as LDL and VLDL, are internalized by CD36 and hydrolyzed via lysosomal acid lipases[105]. Th2 cytokines can activate signal transducer and activator of transcription (STAT) 6 and the down-stream transcriptional regulators, such as peroxisome proliferator-activated receptor (PPAR) β,
Taken together, enhanced glucose uptake and glycolysis are key characteristics of M1 macrophages, whereas elevated fatty acid oxidation and oxidative phosphorylation are the main features of M2 macrophages. As reviewed recently, long chain non-coding RNAs may also participate in the regulation of macrophage polarization[124]. The underlying mechanisms of how metabolism regulates macrophage polarization are complex and remain open[125-127]. However, the development of advanced technologies, such as single-cell resolution and the metabolomics, are sure to improve our knowledge in this area.
MACROPHAGE POLARIZATION INDUCED BY ENDOGENOUS MOLECULES
As described above, macrophages are plastic cells that can polarize to different phenotypes upon different stimulus[128-130]. The polarization of plaque macrophages is mainly determined by the dynamic microenvironment, such as cytokines, lipids, and other molecules, present in atherosclerotic plaques[93,129,131,132]. According to the dichotomy, macrophages are divided into two classes: classically activated pro-inflammatory M1 macrophages and alternatively activated anti-inflammatory M2 macrophages[66,133]. Presently, available data related to macrophage polarization are mainly focused on the switch between M1 and M2 phenotypes.
As described in the previous section, macrophage phenotype can be regulated by cytokines and growth factors present in atherosclerotic plaques[134]. Pro-inflammatory M1 macrophages are typically triggered by T helper 1 (Th1) cytokines and boost an inflammatory response, whereas M2 macrophages are mainly induced by Th2 cytokines and counterbalance inflammation via producing anti-inflammatory cytokines[65,135,136]. IL-4 cytokine could polarize macrophages to the M2a phenotype resident in areas of neovascularization and stable plaques[20,94,133]. Granulocyte-macrophage colony-stimulating factor and macrophage colony-stimulating factor are important growth factors regulating differentiation and polarization of macrophages. GM-CSF induces M1 macrophage through promoting the expression of PPBP (also known as CXCL7) and AKT2/extracellular signal-regulated kinase (ERK)/NF-κB and Janus kinase (JAK) 2/STAT5 pathways, whereas M-CSF induced M2 phenotypes through enhancing PI3K/ERK/SP1 pathway and blocking NF-κB via PI3K/AKT1 pathway[136,137-141]. The dynamic ratio of GM-CSF and M-CSF might determine the macrophage polarization[140,142]. In atherosclerotic plaques, another stimulus for macrophage polarization is intraplaque hemorrhage that occurs after vessel rupture in the areas of neovascularization, as mentioned above[97,98,143].
During the progression of atherosclerosis, deposition of ox-LDL and other oxidized lipoproteins induces M1 polarization through activation of a TLR4-mediated pathway and inhibition of the transcription factor Krüppel-like factor (KLF) 2[144-146]. Cholesterol crystals during the early stages of the atherosclerotic plaques induce polarization of the M1 phenotype through activation of IL-1 and IL-18 by inducing the caspase-1-activated NLRP3 inflammasome[147,148]. Cholesteryl esters, including linoleate and 7-etocholesteryl-9-carboxynonanoate, can also polarize macrophages to M1 phenotype via activation of the TLR-4 and/or
It should be noted that many molecules may participate in macrophage polarization. For example, myeloid DAP12-associating lectin-1 (MDL-1), also called CLEC5A, is positively correlated with the severity of atherosclerosis and M1 macrophages markers. MDL-1 overexpression could maintain macrophage survival, downregulate cleaved caspase-3 expression, and induce MCP-1 production under atherosclerotic microenvironment stimuli (e.g., ox-LDL and hypoxia) during early atherosclerotic plaque progression[160]. A subunit of the Skp-Cullin-F-box ubiquitin E3 ligase device, called FBXO3, is also associated with inflammatory response of macrophages upon ox-LDL stimuli[161]. Of note, galectin-3, also known as Mac-2, is a β-galactoside-binding lectin highly and constitutively expressed on plaque macrophages and displays a soluble form in the tissue microenvironment and plasma[162,163]. This lectin-molecule has been proposed for imaging atherosclerotic lesions in vivo using (89Zr)-DFO-Galectin 3-F(ab')2 mAb PET/CT. This novel method can image atherosclerotic plaques at different stages and has potential application for clinical detection of atherosclerosis[164]. Functionally, galectin-3 acts as an opsonin and enhances macrophage efferocytosis of apoptotic cells[165,166]. In M1 macrophages, galectin-3 endocytosis is receptor mediated and carbohydrate independent, whereas, in M2 macrophages, this process is carbohydrate recognition domain mediated[167]. Furthermore, the expression and secretion of galectin-3 is associated with fibrosis and wound healing[168,169]. A recent study demonstrated that loss of galectin-3 is associated with increased expression of proinflammatory genes, and MMP12-dependent cleavage of galectin-3 may promote proinflammatory macrophage polarization[162]. Furthermore, the IL-10-STAT3-galectin-3 axis plays an important role in regulation of alternative activated macrophages[170,171]. However, previous studies suggested that galectin-3 has both pro- and anti-atherosclerotic roles[172-174]. The modulatory mechanisms of these molecules need to be investigated in the future. Collectively, the atherosclerotic plaque microenvironment impacts the macrophage phenotypes, which in turn affect plaque microenvironment due to their different biological functions and secretion of pro- and anti-inflammatory factors, contributing to atherosclerotic plaque progression and regression[65,175].
SIGNALING PATHWAYS INVOLVED IN MACROPHAGE POLARIZATION
Based on their working mechanisms, the polarization of macrophages is modulated through the activation of transcription factors and signaling pathways, and the receptors respond to different inducers. For example, direct injection of inflammatory proteins is related to the acceleration of atherosclerotic plaque formation by polarization of M1 macrophages in a porcine atherosclerosis model[176]. The key pathways in macrophage polarization are STATs, PI3K/AKT, NF-κB, and MAPKs pathways[177]. It is worth noting that different PI3K/AKT isoforms have different functions in the activation of M1 and M2 macrophage polarization[178-181]. Upon macrophage M1 polarization, MAPKs, STAT-1/2, NF-κB, and other signaling pathways are activated, thereby promoting M1-related genes expression[182,183]. IL-4 induced M2 polarization is associated with the activation of STAT3/6 or PI3K/AKT, which promote M2 genes expression[182-184].
The major pathways that participated in M1 macrophage polarization are summarized in Figure 3. In response to an inflammatory environment, TLR4 is activated, which stimulates intracellular signaling cascades, including MAPK and NF-κB transducers, contributing to the expression of inflammatory cytokines, such as TNF-α, IL-6, and IL-12[185]. IFN-γ-mediated JAK-STAT1/2 signaling also promotes M1 macrophage polarization[183]. Theoretically, molecules that positively regulate these pathways may promote macrophage polarization towards an M1-like phenotype. For example, triggering receptor expressed on myeloid cells (TREM-1) is expressed in macrophages and plays a critical role in the upregulation of CD36, thereby promoting the formation of inflammatory foam cells. Furthermore, TREM-1 promotes inflammation by activating TLR4. Genetic and pharmacological inhibition of TREM-1 reduces TLR4-initiated pro-inflammatory responses and retards the development of atherosclerosis in mice[186]. Proprotein convertase subtillisin/kexin type 9 (PCSK9) contributes to the formation of foam cells by inducing the expression of scavenger receptors and promoting LDLR degradation in lysosome[24]. PCSK9 overexpression in macrophages increases the secretion of pro-inflammatory cytokines induced by ox-LDL in vitro, and the mechanisms are related to the upregulation of TLR4 and p-IκBα levels and NF-κB translocation[187]. PCSK9 gene interference reduces vascular inflammation via inhibiting TLR4/NF-κB signaling pathway[188]. Junction adhesion molecule-like (JAML) protein is expressed in macrophages of human and mouse atherosclerotic plaques. It regulates the activation of NF-κB and the production and secretion of inflammatory cytokines[189]. Silencing the expression of JAML can reduce the progression of atherosclerotic plaques in mouse models[189]. Thyroid-stimulating hormone level is an independent risk factor of atherosclerosis. Ablation of thyroid-stimulating hormone receptor inhibits the formation of atherosclerosis by reducing vascular inflammation and macrophage burden[190]. Mechanistically, thyroid-stimulating hormone interacts with its receptor and activates MAPKs (ERK1/2, p38, and c-Jun N-terminal kinase, JNK) and IκB/p65
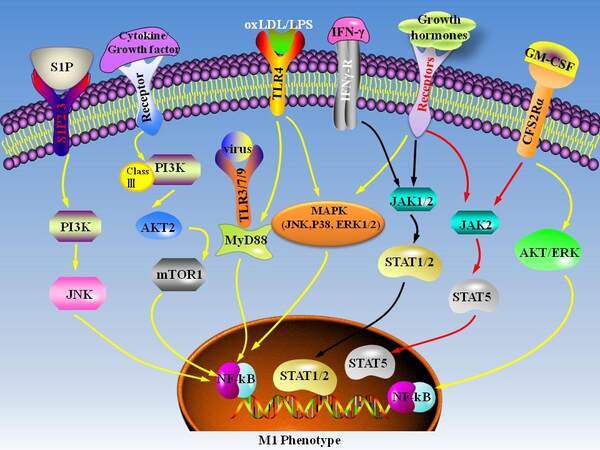
Figure 3. The signaling pathways involved in M1 macrophage polarization. S1P: Sphingosine-1-phosphate; PI3K: phosphoinositide 3-kinase; JNK: c-Jun NH2-terminal kinase; AKT: serine/threonine-protein kinases; mTOR: mechanistic target of rapamycin; TLR: toll-like receptors; MyD88: myeloid differentiation factor 88; OxLDL: oxidized low-density lipoprotein; LPS: lipopolysaccharide; MAPK: mitogen-activated protein kinases; ERK: extracellular regulated protein kinases; IFN-γ: Interferon-gamma; JAK: janus kinase; STAT: signal transducers and activators of transcription; GM-CSF: granulocyte-macrophage colony stimulating factor; NFkB: nuclear factor kappa-B.
The major pathways involved in M2 macrophage polarization are summarized in Figure 4. IL-4- or IL-13-induced IL-4 receptor α signals promote M2 macrophage polarization via the JAK/STAT6 pathway[183,197]. It has been demonstrated that STAT6 facilitates atherosclerotic plaque stabilization by promoting the polarization of macrophages to an M2 subtype and antagonizing ox-LDL-induced cell apoptosis and lipid deposition in a Wnt-β-catenin-dependent manner[177]. KLF4 coordinates with STAT6 to promote M2 genetic program, and it inhibits M1 macrophage polarization via blocking NF-κB pathway[198]. Lack of KLF4 enhances M1 gene expression and promotes pro-inflammatory activation and foam cell formation in vitro[198]. Deletion of KLF4 promotes inflammation and atherosclerotic plaque formation in vivo[199]. Kallistatin, a tissue kallikrein-binding protein and a serine protease inhibitor, reduces atherosclerotic plaque formation in apolipoprotein E-deficient mice. Mechanistically, kallistatin increases the expression of IL-10 and ARG1 that are associated with M2 macrophages and decreases iNOS and monocyte chemotactic protein 1 that are associated with M1 macrophages in a KLF4-dependent manner[200].
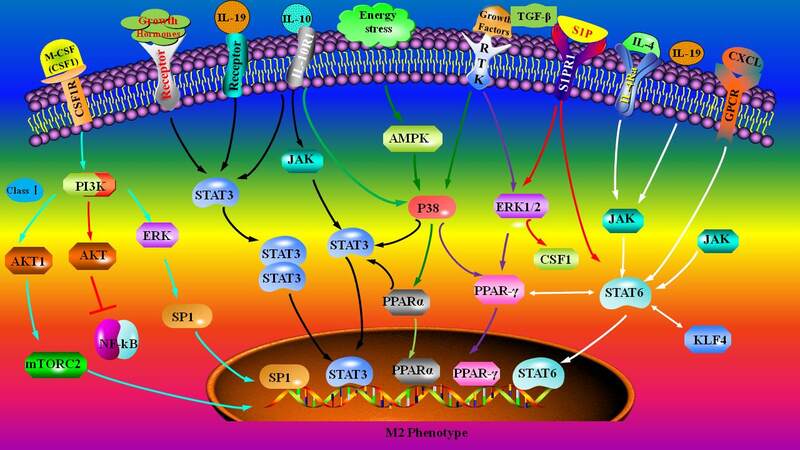
Figure 4. The signaling pathways involved in M2 macrophage polarization. S1P: Sphingosine-1-phosphate; PI3K: phosphoinositide 3-kinase; AKT: serine/threonine-protein kinases; mTOR: mechanistic target of rapamycin; ERK: extracellular regulated protein kinases; JAK: janus kinase; STAT: signal transducers and activators of transcription; M-CSF: macrophage colony stimulating factor; NFkB: nuclear factor kappa-B; SP1: specificity protein 1; IL: interleukin; AMPK: AMP-activated protein kinase; PPAR: peroxisome proliferator-activated receptor; RTK: receptor tyrosine kinase; TGF-β: transforming growth factor-β; CSF1: colony-stimulating factor 1; CXCL: chemokine (C-X-C motif) ligand; GPCR: G-protein-coupled receptor; KLF4: kruppel-like factor 4.
The activation of IL-4 receptor α and other factors may trigger PI3K/AKT signaling, thereby contributing to the M2 macrophage polarization[183,201]. Here again, we need to point out that distinct PI3K/AKT isoforms play distinct roles in activating macrophage polarization. Class I PI3K/AKT1 and Class III PI3K/AKT2 may promote M2 and M1 macrophage polarization, respectively[178-181]. miRNAs also participate in macrophage polarization. For instance, miR-135b promotes inflammation in atherosclerotic plaques, while its inhibition attenuates inflammation via inactivating the PI3K/Akt signaling pathway[202]. Furthermore, activation of PPARγ by its endogenous agonists, such as by fatty acids, can antagonize the activities of the transcription factors STAT and NF-κB, contributing to the anti-inflammatory effect in macrophages[203]. Of note, ligand-dependent SUMOylation of the PPARγ ligand-binding domain is important for PPARγ-mediated suppression of inflammatory response[204]. IL-19 treatment induces a reduction in plaque macrophage numbers and an enrichment in M2 macrophage markers, suggesting an atherosclerosis regression[205]. Mechanistic studies revealed that IL-19 or recombinant IL-19 promotes the activation of the key pathways involved in M2 macrophage polarization, including STAT3/6, KLF4, and PPARγ in vivo[135,205-207]. Additionally, bone morphogenetic proteins, members of the TGF-β superfamily, play a key role in cell fate and differentiation[208,209]. For instance, bone morphogenetic protein-7 directs cellular plasticity, specifically THP-1 monocytes differentiation into M2 macrophages, and enhances the levels of anti-inflammatory cytokines in vitro. Furthermore, this molecule can inhibit plaque formation in vivo[208,210,211].
Finally, ABC transporters also contribute to macrophage polarization towards an M2 phenotype by promoting cholesterol efflux and the subsequent reduction of inflammation. Human atherosclerotic plaques contain a relatively high concentration of desmosterol, which is an endogenous LXR agonist even in the absence of sterol hydroxylases. LXR activation promotes the expression of ABC transporters, thereby enhancing cholesterol efflux accompanied with anti-inflammatory effect. These data suggest that desmosterol might promote macrophage polarization towards an M2 phenotype[212]. miR-23a-5p is positively correlated with plaque progression and vulnerability. Transfection of miR-23a-5p inhibitors can increase the expression of ABCA1/G1 to promote cholesterol efflux and reduce the formation of foam cells[213]. Therefore, lipid-lowering may enhance macrophage polarization towards an M2 phenotype by improving the plaque microenvironment.
TREATMENT DIRECTED MACROPHAGE POLARIZATION
Presently, lipid-lowering strategies cannot completely inhibit atherosclerosis. Macrophages are involved in all stages of this chronic and multifaceted disease. As described above, the dynamic atherosclerotic plaque microenvironment determines the activation states of the macrophages. More importantly, macrophages have a good plasticity and play key roles in plaque instability and regression[214,215]. Therefore, a new idea for the treatment of atherosclerosis is through regulating macrophage polarization from the pro-atherogenic M1 phenotype to the anti-atherogenic M2 phenotype. Theoretically, compounds that regulate the dynamic plaque microenvironment or trigger the signaling pathways involved in macrophage polarization may promote macrophage phenotype shift, contributing to the progression or regression of atherosclerosis.
Melatonin, an indoleamine hormone, shows cardioprotective activity in several animal models[216-220]. Of note, this molecule can inhibit the development of atherosclerosis[221]. Mechanistically, melatonin ameliorates intraplaque inflammation by suppressing macrophage differentiation towards the pro-inflammatory M1 phenotype and improving M2 macrophage polarization as well as RORα-mediated anti-inflammatory effects in vulnerable plaques[222]. It is suggested that the melatonin-RORα axis regulates macrophage polarization by differentially regulating the AMPKα/STATs pathway[222]. The natural product Ginsenoside Rb1 reduces M1 macrophage markers and increases M2 macrophage markers via promoting phosphorylation of STAT6 in vitro. This molecule enhances plaque stability by promoting the anti-inflammatory M2 macrophage polarization in an STAT6-dependent manner in apolipoprotein E-deficient mice[223,224].
Oleoylethanolamide (OEA), an endogenous PPARα agonist, enhances the expression of two classic M2 macrophage markers, CD206 and TGF-β, and decreases the expression of iNOS, an M1 macrophage marker, in THP-1-derived macrophages[225,226]. More importantly, OEA inhibits M1 macrophage polarization in atherosclerotic plaques and promotes the polarization of M2 macrophages through AMPK/PPARα/STAT3 pathway[225,226]. Blocking of PPARα using siRNA or inhibition of AMPK by its inhibitor attenuates the OEA-induced expression of M2 macrophage markers[225,226]. Furthermore, OEA increases collagen content and decreases necrotic core size in atherosclerotic plaques in mice via PPARα[225,226]. Ginsenoside Rg3 (GRg3) has potential vascular protective effects. In vitro study demonstrated that GRg3 reverses the macrophage polarization towards an M1 phenotype induced by advanced glycation end-products. In vivo, it improves the stability of atherosclerotic plaques, reduces M1 macrophages, and increases M2 macrophages through the PPARγ pathway[227]. These results suggest that PPARα or PPARγ agonists have potential effects on promoting M2 macrophage polarization.
Natural products exhibit attractive effects on promoting macrophage polarization towards an M2 phenotype. Curcumin, a polyphenol component in turmeric, has multiple pharmacological activities, including regulation of cholesterol homeostasis, inflammatory response, and especially modulatory effects on macrophage polarization[20,228,229]. In vitro data indicate that curcumin inhibit polarization of RAW264.7 cells (M0) to M1 phenotype and switch M1 or M0 macrophages to the M2 phenotype[228]. Mechanistically, curcumin could suppress M1 inflammatory phenotype through activation of IκBα in M0 macrophages and promote polarization of M1 or M0 phenotypes to M2 macrophages through activation of PPAR-γ[228,229]. Furthermore, accumulating studies demonstrated that curcumin shifts M1 macrophages to an M2 phenotype via downregulating TLR4 and the phosphorylation of MAPKs (ERK, JNK, and p38) and
Some synthesized compounds or clinically used drugs also induce macrophage polarization. Activated factor X contributes to the pathophysiology of chronic inflammation besides its roles in coagulation cascade. Rivaroxaban, an inhibitor of factor X, can inhibit inflammation in the atherosclerotic plaques and in RAW264.7 macrophages except for its antithrombotic effect[235]. Angiotensin 1-7 exhibits cardiovascular protective effect, and its non-peptide mimic AVE0991 can inhibit monocytes/macrophages differentiation towards an M1 phenotype and their recruitment to the perivascular space, thereby retarding inflammation and early atherosclerosis[236]. Dipeptidyl peptidase inhibitors used for treating patients with type 2 diabetes have been found to reduce cholesterol levels and downregulate the formation of atherosclerosis[237,238]. Mechanistically, these inhibitors may promote monocyte polarization towards M2 macrophages through the SDF-1/CXCR4 signaling pathway[238]. Additionally, treatment with RXRα modulator K-80003 decreases 7-ketocholesterol-induced p65 nuclear translocation, IκBα degradation, and transcription of NF-κB target genes, contributing to the reduced plaque size, plaque rupture, macrophage infiltration, and inflammatory cytokine levels[239]. Finally, we should pay attention to some drugs that can promote macrophage polarization towards M1 phenotype. For instance, doxorubicin, a drug used to treat cancers, induces adverse cardiotoxic effects. Mechanistically, doxorubicin treatment increases expression of TLR4 and NLRP3 and downstream MAPK cell signaling proteins, such as MyD88, p-P38, and p-JNK, contributing to the polarization of pro-inflammatory M1 macrophages and secretion of inflammatory cytokines[240]. These drugs need to be carefully used when patients are suffering from cardiovascular disease and cancer at the same time.
CONCLUSIONS AND PERSPECTIVES
Macrophages play important roles in the progression and regression of atherosclerosis, and each subtype of macrophage has distinct functions. These heterogenous and plastic cells can switch from one phenotype to another in response to distinct microenvironmental stimuli, which differentially activate the signaling pathways involved in macrophage polarization. Accumulating data demonstrate that macrophage polarization towards the atheroprotective M2 phenotype may contribute to plaque stability and regression. Theoretically, molecules that positively regulate M2 polarization and/or negatively modulate M1 polarization may benefit atherosclerosis stability and regression. These findings provide a hope for reversing this chronic disease because the present therapeutic strategies cannot completely retard the progression of atherosclerosis. Of note, there are still many questions that need to be resolved to reach this clinical goal.
Macrophages can undergo distinct polarization states, but we do not clearly know the functions of each subtype. At present, the majority of available data are limited to the M1 and M2 phenotypes, which have been simply classified a decade ago. It is worth noting that M2 phenotype macrophages are not always protective, which further increases the complexity of macrophage phenotype and function. Combined proteomic and transcriptomic single-cell analysis is likely to resolve this problem. Furthermore, most studies are carried out in rodents, whose macrophage phenotypes and atherosclerosis progression are different from ours. Therefore, these data cannot be translated directly into clinical application. Novel technologies, such as non-invasive detection, are needed to improve our understanding of human macrophage phenotypic diversity and its roles in atherosclerotic plaques.
DECLARATIONS
Authors’ contributionsPerformed reference collection and wrote the manuscript: Liu N, Zhang B, Sun Y
Re-edited the manuscript: Song W, Guo S
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the Natural Science Foundation of China (81770463, 82070469, 31300639).
Conflicts of interestNot applicable.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
2. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 2013;13:709-21.
3. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res 2016;118:653-67.
4. Amengual J, Barrett TJ. Monocytes and macrophages in atherogenesis. Curr Opin Lipidol 2019;30:401-8.
6. Momtazi-Borojeni AA, Abdollahi E, Nikfar B, Chaichian S, Ekhlasi-Hundrieser M. Curcumin as a potential modulator of M1 and M2 macrophages: new insights in atherosclerosis therapy. Heart Fail Rev 2019;24:399-409.
7. Smith JD, Trogan E, Ginsberg M, Grigaux C, Tian J, Miyata M. Decreased atherosclerosis in mice deficient in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc Natl Acad Sci U S A 1995;92:8264-8.
9. Tsaousi A, Hayes EM, Di Gregoli K, et al. Plaque size is decreased but M1 macrophage polarization and rupture related metalloproteinase expression are maintained after deleting T-Bet in ApoE null mice. PLoS One 2016;11:e0148873.
10. Stöger JL, Gijbels MJ, van der Velden S, et al. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis 2012;225:461-8.
11. Cho KY, Miyoshi H, Kuroda S, et al. The phenotype of infiltrating macrophages influences arteriosclerotic plaque vulnerability in the carotid artery. J Stroke Cerebrovasc Dis 2013;22:910-8.
12. Shaikh S, Brittenden J, Lahiri R, Brown PA, Thies F, Wilson HM. Macrophage subtypes in symptomatic carotid artery and femoral artery plaques. Eur J Vasc Endovasc Surg 2012;44:491-7.
13. Melendez QM, Krishnaji ST, Wooten CJ, Lopez D. Hypercholesterolemia: The role of PCSK9. Arch Biochem Biophys 2017;625-626:39-53.
14. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003;19:71-82.
16. Kim H. The transcription factor MafB promotes anti-inflammatory M2 polarization and cholesterol efflux in macrophages. Sci Rep 2017;7:7591.
17. Solanki S, Dube PR, Birnbaumer L, Vazquez G. Reduced necrosis and content of apoptotic M1 macrophages in advanced atherosclerotic plaques of mice with macrophage-specific loss of Trpc3. Sci Rep 2017;7:42526.
18. Paul A, Lydic TA, Hogan R, Goo YH. Cholesterol acceptors regulate the lipidome of macrophage foam cells. Int J Mol Sci 2019;20:3784.
19. Medbury HJ, Tarran SL, Guiffre AK, Williams MM, Lam TH, et al. Monocytes contribute to the atherosclerotic cap by transformation into fibrocytes. Int Angiol 2008;27:114-23.
20. Bouhlel MA, Derudas B, Rigamonti E, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab 2007;6:137-43.
21. Chistiakov DA, Bobryshev YV, Nikiforov NG, Elizova NV, Sobenin IA, Orekhov AN. Macrophage phenotypic plasticity in atherosclerosis: The associated features and the peculiarities of the expression of inflammatory genes. Int J Cardiol 2015;184:436-45.
22. Adamson S, Leitinger N. Phenotypic modulation of macrophages in response to plaque lipids. Curr Opin Lipidol 2011;22:335-42.
23. Nagenborg J, Goossens P, Biessen EAL, Donners MMPC. Heterogeneity of atherosclerotic plaque macrophage origin, phenotype and functions: Implications for treatment. Eur J Pharmacol 2017;816:14-24.
24. Guo S, Xia X, Gu H, Zhang D. Proprotein convertase subtilisin/kexin-type 9 and lipid metabolism. Adv Exp Med Biol 2020;1276:137-56.
25. She ZG, Hamzah J, Kotamraju VR, Pang HB, Jansen S, Ruoslahti E. Plaque-penetrating peptide inhibits development of hypoxic atherosclerotic plaque. J Control Release 2016;238:212-20.
26. Tabas I, Lichtman AH. Monocyte-macrophages and T cells in atherosclerosis. Immunity 2017;47:621-34.
27. Rahman MS, Murphy AJ, Woollard KJ. Effects of dyslipidaemia on monocyte production and function in cardiovascular disease. Nat Rev Cardiol 2017;14:387-400.
28. Mildner A, Marinkovic G, Jung S. Murine monocytes: origins, subsets, fates, and functions. Microbiol Spectr 2016:4.
29. Jia SJ, Gao KQ, Zhao M. Epigenetic regulation in monocyte/macrophage: A key player during atherosclerosis. Cardiovasc Ther 2017;35:e12262.
30. Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical monocytes in health and disease. Annu Rev Immunol 2019;37:439-56.
32. Xiang GA, Zhang YD, Su CC, et al. Dynamic changes of mononuclear phagocytes in circulating, pulmonary alveolar and interstitial compartments in a mouse model of experimental silicosis. Inhal Toxicol 2016;28:393-402.
33. Hamers AAJ, Dinh HQ, Thomas GD, et al. Human monocyte heterogeneity as revealed by high-dimensional mass cytometry. Arterioscler Thromb Vasc Biol 2019;39:25-36.
34. Villani AC, Satija R, Reynolds G, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017;356:eaah4573.
35. Wildgruber M, Aschenbrenner T, Wendorff H, et al. The "Intermediate" CD14++CD16+ monocyte subset increases in severe peripheral artery disease in humans. Sci Rep 2016;6:39483.
36. Saha AK, Osmulski P, Dallo SF, Gaczynska M, Huang TH, Ramasubramanian AK. Cholesterol regulates monocyte rolling through CD44 distribution. Biophys J 2017;112:1481-8.
37. Tolani S, Pagler TA, Murphy AJ, et al. Hypercholesterolemia and reduced HDL-C promote hematopoietic stem cell proliferation and monocytosis: studies in mice and FH children. Atherosclerosis 2013;229:79-85.
38. Schnitzler JG, Bernelot Moens SJ, Tiessens F, et al. Nile Red Quantifier: a novel and quantitative tool to study lipid accumulation in patient-derived circulating monocytes using confocal microscopy. J Lipid Res 2017;58:2210-9.
39. Christensen JJ, Osnes LT, Halvorsen B, et al. Altered leukocyte distribution under hypercholesterolemia: A cross-sectional study in children with familial hypercholesterolemia. Atherosclerosis 2017;256:67-74.
40. Combadière C, Potteaux S, Rodero M, et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 2008;117:1649-57.
41. Murphy AJ, Tall AR. Disordered haematopoiesis and athero-thrombosis. Eur Heart J 2016;37:1113-21.
42. Fernandez-Ruiz I, Puchalska P, Narasimhulu CA, Sengupta B, Parthasarathy S. Differential lipid metabolism in monocytes and macrophages: influence of cholesterol loading. J Lipid Res 2016;57:574-86.
43. Lian Z, Saeed A, Peng X, et al. Monocyte phenotyping and management of lipoprotein X syndrome. J Clin Lipidol 2020;14:850-8.
44. Bekkering S, Stiekema LCA, Bernelot Moens S, et al. Treatment with Statins does not revert trained immunity in patients with familial hypercholesterolemia. Cell Metab 2019;30:1-2.
45. Li W, Sultana N, Siraj N, et al. Autophagy dysfunction and regulatory cystatin C in macrophage death of atherosclerosis. J Cell Mol Med 2016;20:1664-72.
46. Tjaden K, Adam C, Godfrey R, Hanley PJ, Pardali E, Waltenberger J. Low density lipoprotein interferes with intracellular signaling of monocytes resulting in impaired chemotaxis and enhanced chemokinesis. Int J Cardiol 2018;255:160-5.
47. Bahrami A, Liberale L, Reiner Ž, Carbone F, Montecucco F, Sahebkar A. Inflammatory biomarkers for cardiovascular risk stratification in familial hypercholesterolemia. Rev Physiol Biochem Pharmacol 2020;177:25-52.
48. Wang Y, Dubland JA, Allahverdian S, et al. Smooth muscle cells contribute the majority of foam cells in ApoE (Apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler Thromb Vasc Biol 2019;39:876-87.
49. Rendra E, Riabov V, Mossel DM, Sevastyanova T, Harmsen MC, Kzhyshkowska J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology 2019;224:242-53.
50. Dekker A, Davis FM, Kunkel SL, Gallagher KA. Targeting epigenetic mechanisms in diabetic wound healing. Transl Res 2019;204:39-50.
51. Kulcsar KA, Coleman CM, Beck SE, Frieman MB. Comorbid diabetes results in immune dysregulation and enhanced disease severity following MERS-CoV infection. JCI Insight 2019;4:131774.
52. Codo AC, Davanzo GG, Monteiro LB, et al. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/Glycolysis-dependent axis. Cell Metab 2020;32:498-9.
53. Ikeda Y, Sonoda N, Bachuluun B, Kimura S, Ogawa Y, Inoguchi T. Aberrant activation of bone marrow Ly6C high monocytes in diabetic mice contributes to impaired glucose tolerance. PLoS One 2020;15:e0229401.
54. Barrett TJ, Distel E, Murphy AJ, et al. Apolipoprotein AI promotes atherosclerosis regression in diabetic mice by suppressing myelopoiesis and plaque inflammation. Circulation 2019;140:1170-84.
55. Csordas A, Bernhard D. The biology behind the atherothrombotic effects of cigarette smoke. Nat Rev Cardiol 2013;10:219-30.
56. Mehta S, Dhawan V. Exposure of cigarette smoke condensate activates NLRP3 inflammasome in THP-1 cells in a stage-specific manner: An underlying role of innate immunity in atherosclerosis. Cell Signal 2020;72:109645.
57. Kohashi K, Nakagomi A, Morisawa T, et al. Effect of smoking status on monocyte tissue factor activity, carotid atherosclerosis and long-term prognosis in metabolic syndrome. Circ J 2018;82:1418-27.
58. Cheng YC, Sheen JM, Hu WL, Hung YC. Polyphenols and oxidative stress in atherosclerosis-related ischemic heart disease and stroke. Oxid Med Cell Longev 2017;2017:8526438.
59. de Ronde MWJ, Kok MGM, Moerland PD, et al. High miR-124-3p expression identifies smoking individuals susceptible to atherosclerosis. Atherosclerosis 2017;263:377-84.
60. Mehta S, Srivastava N, Bhatia A, Dhawan V. Exposure of cigarette smoke condensate activates NLRP3 inflammasome in vitro and in vivo: A connotation of innate immunity and atherosclerosis. Int Immunopharmacol 2020;84:106561.
61. Studer RK, Negrete H, Craven PA, DeRubertis FR. Protein kinase C signals thromboxane induced increases in fibronectin synthesis and TGF-beta bioactivity in mesangial cells. Kidney Int 1995;48:422-30.
62. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 2004;25:677-86.
63. Boldrick JC, Alizadeh AA, Diehn M, et al. Stereotyped and specific gene expression programs in human innate immune responses to bacteria. Proc Natl Acad Sci U S A 2002;99:972-7.
64. Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science 1993;259:1739-42.
66. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011;11:723-37.
67. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol 2014;5:614.
68. Yang R, Liao Y, Wang L, et al. Exosomes derived from M2b macrophages attenuate DSS-induced colitis. Front Immunol 2019;10:2346.
69. Delavary B, van der Veer WM, van Egmond M, Niessen FB, Beelen RH. Macrophages in skin injury and repair. Immunobiology 2011;216:753-62.
70. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014;17:109-18.
71. Lee CG, Homer RJ, Zhu Z, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med 2001;194:809-21.
72. Spencer M, Yao-Borengasser A, Unal R, et al. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. Am J Physiol Endocrinol Metab 2010;299:E1016-27.
73. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008;8:958-69.
74. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci 2008;13:453-61.
75. Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol 2012;189:3508-20.
76. Zizzo G, Cohen PL. IL-17 stimulates differentiation of human anti-inflammatory macrophages and phagocytosis of apoptotic neutrophils in response to IL-10 and glucocorticoids. J Immunol 2013;190:5237-46.
77. Ferrante CJ, Pinhal-Enfield G, Elson G, et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation 2013;36:921-31.
78. Grinberg S, Hasko G, Wu D, Leibovich SJ. Suppression of PLCbeta2 by endotoxin plays a role in the adenosine A(2A) receptor-mediated switch of macrophages from an inflammatory to an angiogenic phenotype. Am J Pathol 2009;175:2439-53.
79. Xu H, Jiang J, Chen W, Li W, Chen Z. Vascular macrophages in atherosclerosis. J Immunol Res 2019;2019:4354786.
80. Pourcet B, Staels B. Alternative macrophages in atherosclerosis: not always protective! J Clin Invest 2018;128:910-2.
81. Kadl A, Meher AK, Sharma PR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res 2010;107:737-46.
82. Finn AV, Nakano M, Polavarapu R, et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol 2012;59:166-77.
83. Boyle JJ, Johns M, Kampfer T, et al. Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ Res 2012;110:20-33.
84. Nielsen MJ, Møller HJ, Moestrup SK. Hemoglobin and heme scavenger receptors. Antioxid Redox Signal 2010;12:261-73.
85. Philippidis P, Mason JC, Evans BJ, et al. Hemoglobin scavenger receptor CD163 mediates interleukin-10 release and heme oxygenase-1 synthesis: antiinflammatory monocyte-macrophage responses in vitro, in resolving skin blisters in vivo, and after cardiopulmonary bypass surgery. Circ Res 2004;94:119-26.
86. Landis RC, Philippidis P, Domin J, Boyle JJ, Haskard DO. Haptoglobin genotype-dependent anti-inflammatory signaling in CD163(+) macrophages. Int J Inflam 2013;2013:980327.
87. Boyle JJ. Heme and haemoglobin direct macrophage Mhem phenotype and counter foam cell formation in areas of intraplaque haemorrhage. Curr Opin Lipidol 2012;23:453-61.
88. Kockx MM, Cromheeke KM, Knaapen MW, et al. Phagocytosis and macrophage activation associated with hemorrhagic microvessels in human atherosclerosis. Arterioscler Thromb Vasc Biol 2003;23:440-6.
89. Boyle JJ, Johns M, Lo J, et al. Heme induces heme oxygenase 1 via Nrf2: role in the homeostatic macrophage response to intraplaque hemorrhage. Arterioscler Thromb Vasc Biol 2011;31:2685-91.
90. Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol 2010;184:4810-8.
91. Barlis P, Serruys PW, Devries A, Regar E. Optical coherence tomography assessment of vulnerable plaque rupture: predilection for the plaque 'shoulder'. Eur Heart J 2008;29:2023.
92. Seneviratne A, Hulsmans M, Holvoet P, Monaco C. Biomechanical factors and macrophages in plaque stability. Cardiovasc Res 2013;99:284-93.
93. Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol 2009;29:1419-23.
94. Chinetti-Gbaguidi G, Baron M, Bouhlel MA, et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARγ and LXRα pathways. Circ Res 2011;108:985-95.
95. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 2010;10:36-46.
96. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 2009;27:451-83.
97. Sindrilaru A, Peters T, Wieschalka S, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest 2011;121:985-97.
98. Bories G, Colin S, Vanhoutte J, et al. Liver X receptor activation stimulates iron export in human alternative macrophages. Circ Res 2013;113:1196-205.
99. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016;354:472-7.
100. Newby AC. Metalloproteinase production from macrophages - a perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp Physiol 2016;101:1327-37.
101. Shioi A, Ikari Y. Plaque calcification during atherosclerosis progression and regression. J Atheroscler Thromb 2018;25:294-303.
102. Reith S, Milzi A, Dettori R, Marx N, Burgmaier M. Predictors for target lesion microcalcifications in patients with stable coronary artery disease: an optical coherence tomography study. Clin Res Cardiol 2018;107:763-71.
103. Burgmaier M, Milzi A, Dettori R, Burgmaier K, Marx N, Reith S. Co-localization of plaque macrophages with calcification is associated with a more vulnerable plaque phenotype and a greater calcification burden in coronary target segments as determined by OCT. PLoS One 2018;13:e0205984.
104. Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol 2017;8:289.
105. Mehla K, Singh PK. Metabolic regulation of macrophage polarization in cancer. Trends Cancer 2019;5:822-34.
106. Qing J, Zhang Z, Novák P, Zhao G, Yin K. Mitochondrial metabolism in regulating macrophage polarization: an emerging regulator of metabolic inflammatory diseases. Acta Biochim Biophys Sin (Shanghai) 2020;52:917-26.
107. Verdeguer F, Aouadi M. Macrophage heterogeneity and energy metabolism. Exp Cell Res 2017;360:35-40.
108. Haschemi A, Kosma P, Gille L, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab 2012;15:813-26.
109. Galván-Peña S, O'Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol 2014;5:420.
110. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019;574:575-80.
111. Wang F, Zhang S, Vuckovic I, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab 2018;28:463-475.e4.
112. Palmieri EM, Gonzalez-Cotto M, Baseler WA, et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat Commun 2020;11:698.
113. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013;496:238-42.
114. Rath M, Müller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol 2014;5:532.
115. Seim GL, Britt EC, John SV, et al. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-γ stimulation. Nat Metab 2019;1:731-42.
116. Ménégaut L, Thomas C, Lagrost L, Masson D. Fatty acid metabolism in macrophages: a target in cardio-metabolic diseases. Curr Opin Lipidol 2017;28:19-26.
117. Rodriguez AE, Ducker GS, Billingham LK, et al. Serine Metabolism Supports Macrophage IL-1β Production. Cell Metab 2019;29:1003-1011.e4.
118. Kieler M, Hofmann M, Schabbauer G. More than just protein building blocks: how amino acids and related metabolic pathways fuel macrophage polarization. FEBS J 2021;288:3694-714.
119. Esteban-Martinez L, Boya P. BNIP3L/NIX-dependent mitophagy regulates cell differentiation via metabolic reprogramming. Autophagy 2018;14:915-17.
120. Wei Y, Corbalán-Campos J, Gurung R, et al. Dicer in Macrophages Prevents Atherosclerosis by Promoting Mitochondrial Oxidative Metabolism. Circulation 2018;138:2007-20.
121. den Bossche J, Baardman J, de Winther MP. Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. J Vis Exp 2015; doi: 10.3791/53424.
122. Vats D, Mukundan L, Odegaard JI, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 2006;4:13-24.
123. Liu PS, Wang H, Li X, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 2017;18:985-94.
124. Simion V, Haemmig S, Feinberg MW. LncRNAs in vascular biology and disease. Vascul Pharmacol 2019;114:145-56.
125. Bonacina F, Da Dalt L, Catapano AL, Norata GD. Metabolic adaptations of cells at the vascular-immune interface during atherosclerosis. Mol Aspects Med 2021;77:100918.
126. Hobson-Gutierrez SA, Carmona-Fontaine C. The metabolic axis of macrophage and immune cell polarization. Dis Model Mech 2018;11:dmm034462.
127. Thapa B, Lee K. Metabolic influence on macrophage polarization and pathogenesis. BMB Rep 2019;52:360-72.
128. Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage polarization and plasticity in health and disease. Immunol Res 2012;53:11-24.
129. Porcheray F, Viaud S, Rimaniol AC, et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol 2005;142:481-9.
130. Lee S, Huen S, Nishio H, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 2011;22:317-26.
131. Abumaree MH, Al Jumah MA, Kalionis B, et al. Human placental mesenchymal stem cells (pMSCs) play a role as immune suppressive cells by shifting macrophage differentiation from inflammatory M1 to anti-inflammatory M2 macrophages. Stem Cell Rev Rep 2013;9:620-41.
132. Nahrendorf M, Swirski FK. Abandoning M1/M2 for a network model of macrophage function. Circ Res 2016;119:414-7.
134. Wolfs IM, Donners MM, de Winther MP. Differentiation factors and cytokines in the atherosclerotic plaque micro-environment as a trigger for macrophage polarisation. Thromb Haemost 2011;106:763-71.
135. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012;122:787-95.
136. Verreck FA, de Boer T, Langenberg DM, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci U S A 2004;101:4560-5.
138. Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol 2014;6:a021857-a021857.
139. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 2014;6:13.
140. Brochériou I, Maouche S, Durand H, et al. Antagonistic regulation of macrophage phenotype by M-CSF and GM-CSF: implication in atherosclerosis. Atherosclerosis 2011;214:316-24.
141. Waldo SW, Li Y, Buono C, et al. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am J Pathol 2008;172:1112-26.
142. Plenz G, Koenig C, Severs NJ, Robenek H. Smooth muscle cells express granulocyte-macrophage colony-stimulating factor in the undiseased and atherosclerotic human coronary artery. Arterioscler Thromb Vasc Biol 1997;17:2489-99.
143. Kolodgie FD, Gold HK, Burke AP, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med 2003;349:2316-25.
144. Bae YS, Lee JH, Choi SH, et al. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ Res 2009;104:210-8, 21p following 218.
145. Tits LJ, Stienstra R, van Lent PL, Netea MG, Joosten LA, Stalenhoef AF. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: a crucial role for Krüppel-like factor 2. Atherosclerosis 2011;214:345-9.
146. Hirose K, Iwabuchi K, Shimada K, et al. Different responses to oxidized low-density lipoproteins in human polarized macrophages. Lipids Health Dis 2011;10:1.
147. Fan A, Wu X, Wu H, et al. Atheroprotective effect of oleoylethanolamide (OEA) targeting oxidized LDL. PLoS One 2014;9:e85337.
148. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010;464:1357-61.
149. Fang L, Harkewicz R, Hartvigsen K, et al. Oxidized cholesteryl esters and phospholipids in zebrafish larvae fed a high cholesterol diet: macrophage binding and activation. J Biol Chem 2010;285:32343-51.
150. Sottero B, Gamba P, Longhi M, et al. Expression and synthesis of TGFbeta1 is induced in macrophages by 9-oxononanoyl cholesterol, a major cholesteryl ester oxidation product. Biofactors 2005;24:209-16.
151. Yang J, Yang L, Tian L, Ji X, Yang L, Li L. Sphingosine 1-phosphate (S1P)/S1P receptor2/3 axis promotes inflammatory M1 polarization of bone marrow-derived monocyte/macrophage via G(α)i/o/PI3K/JNK pathway. Cell Physiol Biochem 2018;49:1677-93.
152. Hou L, Yang L, Chang N, et al. Macrophage sphingosine 1-phosphate receptor 2 blockade attenuates liver inflammation and fibrogenesis triggered by NLRP3 inflammasome. Front Immunol 2020;11:1149.
153. Mitchell PL, McLeod RS. Conjugated linoleic acid and atherosclerosis: studies in animal models. Biochem Cell Biol 2008;86:293-301.
154. McCarthy C, Duffy MM, Mooney D, et al. IL-10 mediates the immunoregulatory response in conjugated linoleic acid-induced regression of atherosclerosis. FASEB J 2013;27:499-510.
155. Müller J, von Bernstorff W, Heidecke CD, Schulze T. Differential S1P receptor profiles on M1- and M2-polarized macrophages affect macrophage cytokine production and Migration. Biomed Res Int 2017;2017:7584621.
156. Park SJ, Lee KP, Kang S, et al. Sphingosine 1-phosphate induced anti-atherogenic and atheroprotective M2 macrophage polarization through IL-4. Cell Signal 2014;26:2249-58.
157. Kuang Y, Li X, Liu X, et al. Vascular endothelial S1pr1 ameliorates adverse cardiac remodelling via stimulating reparative macrophage proliferation after myocardial infarction. Cardiovasc Res 2021;117:585-99.
158. Titos E, Rius B, González-Périz A, et al. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol 2011;187:5408-18.
159. Serhan CN, Yang R, Martinod K, et al. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med 2009;206:15-23.
160. Xiong W, Wang H, Lu L, et al. The macrophage C-type lectin receptor CLEC5A (MDL-1) expression is associated with early plaque progression and promotes macrophage survival. J Transl Med 2017;15:234.
161. Chandra D, Londino J, Alexander S, et al. The SCFFBXO3 ubiquitin E3 ligase regulates inflammation in atherosclerosis. J Mol Cell Cardiol 2019;126:50-9.
162. Di Gregoli K, Somerville M, Bianco R, et al. Galectin-3 identifies a subset of macrophages with a potential beneficial role in atherosclerosis. Arterioscler Thromb Vasc Biol 2020;40:1491-509.
163. Falcone C, Lucibello S, Mazzucchelli I, et al. Galectin-3 plasma levels and coronary artery disease: a new possible biomarker of acute coronary syndrome. Int J Immunopathol Pharmacol 2011;24:905-13.
164. Varasteh Z, De Rose F, Mohanta S, et al. Imaging atherosclerotic plaques by targeting Galectin-3 and activated macrophages using (89Zr)-DFO- Galectin3-F(ab')2 mAb. Theranostics 2021;11:1864-76.
165. Karlsson A, Christenson K, Matlak M, et al. Galectin-3 functions as an opsonin and enhances the macrophage clearance of apoptotic neutrophils. Glycobiology 2009;19:16-20.
166. Erriah M, Pabreja K, Fricker M, et al. Galectin-3 enhances monocyte-derived macrophage efferocytosis of apoptotic granulocytes in asthma. Respir Res 2019;20:1.
167. Lepur A, Carlsson MC, Novak R, Dumić J, Nilsson UJ, Leffler H. Galectin-3 endocytosis by carbohydrate independent and dependent pathways in different macrophage like cell types. Biochim Biophys Acta 2012;1820:804-18.
168. Henderson NC, Mackinnon AC, Farnworth SL, et al. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am J Pathol 2008;172:288-98.
169. Cassaglia P, Penas F, Betazza C, et al. Genetic deletion of galectin-3 alters the temporal evolution of macrophage infiltration and healing affecting the cardiac remodeling and function after myocardial infarction in mice. Am J Pathol 2020;190:1789-800.
170. MacKinnon AC, Farnworth SL, Hodkinson PS, et al. Regulation of alternative macrophage activation by galectin-3. J Immunol 2008;180:2650-8.
171. Shirakawa K, Endo J, Kataoka M, et al. IL (Interleukin)-10-STAT3-Galectin-3 axis is essential for osteopontin-producing reparative macrophage polarization after myocardial infarction. Circulation 2018;138:2021-35.
172. Iacobini C, Menini S, Ricci C, et al. Accelerated lipid-induced atherogenesis in galectin-3-deficient mice: role of lipoxidation via receptor-mediated mechanisms. Arterioscler Thromb Vasc Biol 2009;29:831-6.
173. Gleissner CA, Erbel C, Linden F, et al. Galectin-3 binding protein, coronary artery disease and cardiovascular mortality: Insights from the LURIC study. Atherosclerosis 2017;260:121-9.
174. Gao Z, Liu Z, Wang R, Zheng Y, Li H, Yang L. Galectin-3 is a potential mediator for atherosclerosis. J Immunol Res 2020;2020:5284728.
175. Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol 2015;12:10-7.
176. Lee SG, Oh J, Bong SK, et al. Macrophage polarization and acceleration of atherosclerotic plaques in a swine model. PLoS One 2018;13:e0193005.
177. Gong M, Zhuo X, Ma A. STAT6 Upregulation promotes M2 macrophage polarization to suppress atherosclerosis. Med Sci Monit Basic Res 2017;23:240-9.
178. Zhai C, Cheng J, Mujahid H, et al. Selective inhibition of PI3K/Akt/mTOR signaling pathway regulates autophagy of macrophage and vulnerability of atherosclerotic plaque. PLoS One 2014;9:e90563.
179. Yan H, Ma Y, Li Y, et al. Insulin inhibits inflammation and promotes atherosclerotic plaque stability via PI3K-Akt pathway activation. Immunol Lett 2016;170:7-14.
180. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol 2017;198:1006-14.
181. Linton MF, Moslehi JJ, Babaev VR. Akt Signaling in Macrophage Polarization, Survival, and Atherosclerosis. Int J Mol Sci 2019;20:2703.
182. Xu R, Li C, Wu Y, et al. Role of KCa3.1 Channels in macrophage polarization and its relevance in atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol 2017;37:226-36.
183. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 2011;11:750-61.
184. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 2010;11:889-96.
185. Bosisio D, Polentarutti N, Sironi M, et al. Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-gamma: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood 2002;99:3427-31.
186. Joffre J, Potteaux S, Zeboudj L, et al. Genetic and pharmacological inhibition of TREM-1 limits the development of experimental atherosclerosis. J Am Coll Cardiol 2016;68:2776-93.
187. Yurtseven E, Ural D, Baysal K, Tokgözoğlu L. An update on the role of PCSK9 in atherosclerosis. J Atheroscler Thromb 2020;27:909-18.
188. Tang ZH, Peng J, Ren Z, et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis 2017;262:113-22.
189. Sokeechand BSH, Trigatti BL. Un-JAMming atherosclerotic arteries: JAM-L as a target to attenuate plaque development. Clin Sci (Lond) 2019;133:1581-5.
190. Yang C, Lu M, Chen W, et al. Thyrotropin aggravates atherosclerosis by promoting macrophage inflammation in plaques. J Exp Med 2019;216:1182-98.
191. Hamers AA, Vos M, Rassam F, et al. Bone marrow-specific deficiency of nuclear receptor Nur77 enhances atherosclerosis. Circ Res 2012;110:428-38.
192. Hanna RN, Shaked I, Hubbeling HG, et al. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res 2012;110:416-27.
193. Kahles F, Liberman A, Halim C, et al. The incretin hormone GIP is upregulated in patients with atherosclerosis and stabilizes plaques in ApoE-/- mice by blocking monocyte/macrophage activation. Mol Metab 2018;14:150-7.
194. Ji Y, Liu J, Wang Z, Li Z. PPARγ agonist rosiglitazone ameliorates LPS-induced inflammation in vascular smooth muscle cells via the TLR4/TRIF/IRF3/IP-10 signaling pathway. Cytokine 2011;55:409-19.
195. Wen H, Liu M, Liu Z, et al. PEDF improves atherosclerotic plaque stability by inhibiting macrophage inflammation response. Int J Cardiol 2017;235:37-41.
196. Yang SL, Chen SL, Wu JY, Ho TC, Tsao YP. Pigment epithelium-derived factor induces interleukin-10 expression in human macrophages by induction of PPAR gamma. Life Sci 2010;87:26-35.
197. Takeda K, Tanaka T, Shi W, et al. Essential role of Stat6 in IL-4 signalling. Nature 1996;380:627-30.
198. Liao X, Sharma N, Kapadia F, et al. Krüppel-like factor 4 regulates macrophage polarization. J Clin Invest 2011;121:2736-49.
199. Sharma N, Lu Y, Zhou G, et al. Myeloid Krüppel-like factor 4 deficiency augments atherogenesis in ApoE-/- mice--brief report. Arterioscler Thromb Vasc Biol 2012;32:2836-8.
200. Li B, Sheng Z, Liu C, et al. Kallistatin inhibits atherosclerotic inflammation by regulating macrophage polarization. Hum Gene Ther 2019;30:339-51.
201. Fruman DA, Snapper SB, Yballe CM, et al. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science 1999;283:393-7.
202. Wu BW, Liu Y, Wu MS, et al. Downregulation of microRNA-135b promotes atherosclerotic plaque stabilization in atherosclerotic mice by upregulating erythropoietin receptor. IUBMB Life 2020;72:198-213.
203. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998;391:79-82.
204. Pascual G, Fong AL, Ogawa S, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 2005;437:759-63.
205. Gabunia K, Ellison S, Kelemen S, et al. IL-19 halts progression of atherosclerotic plaque, polarizes, and increases cholesterol uptake and efflux in macrophages. Am J Pathol 2016;186:1361-74.
206. Feig JE, Parathath S, Rong JX, et al. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation 2011;123:989-98.
207. Khallou-Laschet J, Varthaman A, Fornasa G, et al. Macrophage plasticity in experimental atherosclerosis. PLoS One 2010;5:e8852.
208. Rocher C, Singla R, Singal PK, Parthasarathy S, Singla DK. Bone morphogenetic protein 7 polarizes THP-1 cells into M2 macrophages. Can J Physiol Pharmacol 2012;90:947-51.
209. Boon MR, van der Horst G, van der Pluijm G, Tamsma JT, Smit JW, Rensen PC. Bone morphogenetic protein 7: a broad-spectrum growth factor with multiple target therapeutic potency. Cytokine Growth Factor Rev 2011;22:221-9.
210. Singla DK, Singla R, Wang J. BMP-7 treatment increases M2 macrophage differentiation and reduces inflammation and plaque formation in Apo E-/- mice. PLoS One 2016;11:e0147897.
211. Rocher C, Singla DK. SMAD-PI3K-Akt-mTOR pathway mediates BMP-7 polarization of monocytes into M2 macrophages. PLoS One 2013;8:e84009.
212. Agil A, Rosado I, Ruiz R, Figueroa A, Zen N, Fernández-Vázquez G. Melatonin improves glucose homeostasis in young Zucker diabetic fatty rats. J Pineal Res 2012;52:203-10.
213. Yang S, Ye ZM, Chen S, et al. MicroRNA-23a-5p promotes atherosclerotic plaque progression and vulnerability by repressing ATP-binding cassette transporter A1/G1 in macrophages. J Mol Cell Cardiol 2018;123:139-49.
214. Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler Thromb Vasc Biol 2013;33:1120-6.
215. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res 2014;114:1852-66.
216. Zhao Y, Xu L, Ding S, et al. Novel protective role of the circadian nuclear receptor retinoic acid-related orphan receptor-α in diabetic cardiomyopathy. J Pineal Res 2017;62:e12378.
217. Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ, Chen Y. Protective role of melatonin in cardiac ischemia-reperfusion injury: From pathogenesis to targeted therapy. J Pineal Res 2018;64:e12471.
218. He B, Zhao Y, Xu L, et al. The nuclear melatonin receptor RORα is a novel endogenous defender against myocardial ischemia/reperfusion injury. J Pineal Res 2016;60:313-26.
219. Zhai M, Liu Z, Zhang B, et al. Melatonin protects against the pathological cardiac hypertrophy induced by transverse aortic constriction through activating PGC-1β: In vivo and in vitro studies. J Pineal Res 2017;63:e12433.
220. Xu L, Su Y, Zhao Y, et al. Melatonin differentially regulates pathological and physiological cardiac hypertrophy: Crucial role of circadian nuclear receptor RORα signaling. J Pineal Res 2019;67:e12579.
221. Ma S, Chen J, Feng J, et al. Melatonin ameliorates the progression of atherosclerosis via mitophagy activation and NLRP3 inflammasome inhibition. Oxid Med Cell Longev 2018;2018:9286458.
222. Ding S, Lin N, Sheng X, et al. Melatonin stabilizes rupture-prone vulnerable plaques via regulating macrophage polarization in a nuclear circadian receptor RORα-dependent manner. J Pineal Res 2019;67:e12581.
223. Qiao L, Zhang X, Liu M, et al. Ginsenoside Rb1 enhances atherosclerotic plaque stability by improving autophagy and lipid metabolism in macrophage foam cells. Front Pharmacol 2017;8:727.
224. Zhang X, Liu MH, Qiao L, et al. Ginsenoside Rb1 enhances atherosclerotic plaque stability by skewing macrophages to the M2 phenotype. J Cell Mol Med 2018;22:409-16.
225. Chen Z, Zhuo R, Zhao Y, et al. Oleoylethanolamide stabilizes atherosclerotic plaque through regulating macrophage polarization via AMPK-PPARα pathway. Biochem Biophys Res Commun 2020;524:308-16.
226. Zhao Y, Yan L, Peng L, et al. Oleoylethanolamide alleviates macrophage formation via AMPK/PPARα/STAT3 pathway. Pharmacol Rep 2018;70:1185-94.
227. Guo M, Xiao J, Sheng X, et al. Ginsenoside Rg3 mitigates atherosclerosis progression in diabetic apoE-/- mice by skewing macrophages to the M2 phenotype. Front Pharmacol 2018;9:464.
228. Chen F, Guo N, Cao G, Zhou J, Yuan Z. Molecular analysis of curcumin-induced polarization of murine RAW264.7 macrophages. J Cardiovasc Pharmacol 2014;63:544-52.
229. Jacob A, Wu R, Zhou M, Wang P. Mechanism of the anti-inflammatory effect of curcumin: PPAR-gamma activation. PPAR Res 2007;2007:89369.
230. Meng Z, Yan C, Deng Q, Gao DF, Niu XL. Curcumin inhibits LPS-induced inflammation in rat vascular smooth muscle cells in vitro via ROS-relative TLR4-MAPK/NF-κB pathways. Acta Pharmacol Sin 2013;34:901-11.
231. Zhou Y, Zhang T, Wang X, et al. Curcumin modulates macrophage polarization through the inhibition of the toll-like receptor 4 expression and its signaling pathways. Cell Physiol Biochem 2015;36:631-41.
232. Cao J, Han Z, Tian L, et al. Curcumin inhibits EMMPRIN and MMP-9 expression through AMPK-MAPK and PKC signaling in PMA induced macrophages. J Transl Med 2014;12:266.
233. Aharoni S, Lati Y, Aviram M, Fuhrman B. Pomegranate juice polyphenols induce a phenotypic switch in macrophage polarization favoring a M2 anti-inflammatory state. Biofactors 2015;41:44-51.
234. Wang N, Zhang X, Ma Z, et al. Combination of tanshinone IIA and astragaloside IV attenuate atherosclerotic plaque vulnerability in ApoE(-/-) mice by activating PI3K/AKT signaling and suppressing TRL4/NF-κB signaling. Biomed Pharmacother 2020;123:109729.
235. Hara T, Fukuda D, Tanaka K, et al. Rivaroxaban, a novel oral anticoagulant, attenuates atherosclerotic plaque progression and destabilization in ApoE-deficient mice. Atherosclerosis 2015;242:639-46.
236. Skiba DS, Nosalski R, Mikolajczyk TP, et al. Anti-atherosclerotic effect of the angiotensin 1-7 mimetic AVE0991 is mediated by inhibition of perivascular and plaque inflammation in early atherosclerosis. Br J Pharmacol 2017;174:4055-69.
237. Derosa G, Ragonesi PD, Fogari E, et al. Sitagliptin added to previously taken antidiabetic agents on insulin resistance and lipid profile: a 2-year study evaluation. Fundam Clin Pharmacol 2014;28:221-9.
238. Brenner C, Franz WM, Kühlenthal S, et al. DPP-4 inhibition ameliorates atherosclerosis by priming monocytes into M2 macrophages. Int J Cardiol 2015;199:163-9.
239. Shen L, Sun Z, Nie P, et al. Sulindac-derived retinoid X receptor-α modulator attenuates atherosclerotic plaque progression and destabilization in ApoE-/- mice. Br J Pharmacol 2019;176:2559-72.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Liu N, Zhang B, Sun Y, Song W, Guo S. Macrophage origin, phenotypic diversity, and modulatory signaling pathways in the atherosclerotic plaque microenvironment. Vessel Plus 2021;5:43. http://dx.doi.org/10.20517/2574-1209.2021.25
AMA Style
Liu N, Zhang B, Sun Y, Song W, Guo S. Macrophage origin, phenotypic diversity, and modulatory signaling pathways in the atherosclerotic plaque microenvironment. Vessel Plus. 2021; 5: 43. http://dx.doi.org/10.20517/2574-1209.2021.25
Chicago/Turabian Style
Liu, Na, Baihui Zhang, Yanlong Sun, Weiguo Song, Shoudong Guo. 2021. "Macrophage origin, phenotypic diversity, and modulatory signaling pathways in the atherosclerotic plaque microenvironment" Vessel Plus. 5: 43. http://dx.doi.org/10.20517/2574-1209.2021.25
ACS Style
Liu, N.; Zhang B.; Sun Y.; Song W.; Guo S. Macrophage origin, phenotypic diversity, and modulatory signaling pathways in the atherosclerotic plaque microenvironment. Vessel Plus. 2021, 5, 43. http://dx.doi.org/10.20517/2574-1209.2021.25
About This Article
Copyright

Data & Comments
Data


 Cite This Article 19 clicks
Cite This Article 19 clicks

 Like This Article 31
likes
Like This Article 31
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.