
The interplay of oxidative stress and ARMS2-HTRA1 genetic risk in neovascular AMD

Abstract
Age-related macular degeneration (AMD) is the leading cause of vision loss in adults over 60 years old globally. There are two forms of advanced AMD: “dry” and “wet”. Dry AMD is characterized by geographic atrophy of the retinal pigment epithelium and overlying photoreceptors in the macular region; whereas wet AMD is characterized by vascular penetrance from the choroid into the retina, known as choroidal neovascularization (CNV). Both phenotypes eventually lead to loss of central vision. The pathogenesis of AMD involves the interplay of genetic polymorphisms and environmental risk factors, many of which elevate retinal oxidative stress. Excess reactive oxygen species react with cellular macromolecules, forming oxidation-modified byproducts that elicit chronic inflammation and promote CNV. Additionally, genome-wide association studies have identified several genetic variants in the age-related maculopathy susceptibility 2/high-temperature requirement A serine peptidase 1 (ARMS2-HTRA1) locus associated with the progression of late-stage AMD, especially the wet subtype. In this review, we will focus on the interplay of oxidative stress and HTRA1 in drusen deposition, chronic inflammation, and chronic angiogenesis. We aim to present a multifactorial model of wet AMD progression, supporting HTRA1 as a novel therapeutic target upstream of vascular endothelial growth factor (VEGF), the conventional target in AMD therapeutics. By inhibiting HTRA1’s proteolytic activity, we can reduce pro-angiogenic signaling and prevent proteolytic breakdown of the blood-retina barrier. The anti-HTRA1 approach offers a promising alternative treatment option to wet AMD, complementary to anti-VEGF therapy.
Keywords
Introduction
Age-related macular degeneration (AMD) is the leading cause of irreversible central vision loss in people over 60 years old[1] and is a burgeoning public health problem in many developed countries due to aging populations[2]. There are two types of AMD: dry AMD, characterized by geographic cellular atrophy in the macula; and wet AMD, a proliferative retinopathy caused by aberrant blood vessel growth from the choroid into the neural retina[3]. While wet AMD only accounts for 10%-15% of patients with AMD, it is responsible for 90% of AMD-related severe visual impairment[4]. Despite recent advances in understanding the mechanisms of AMD progression, it remains unclear what predisposes individuals to develop dry vs. wet AMD and what triggers the transition from dry to wet AMD in certain patients. Finding answers to these questions will not only catalyze the development of more effective therapeutics against AMD but may also elucidate mechanisms underlying other neovascular diseases.
Like many age-related diseases, AMD is believed to result from an interplay of genetic, environmental, and behavioral risk factors. The complex etiology of AMD closely resembles that of polypoidal choroidal vasculopathy (PCV) compared to other retinopathies [Table 1]. However, the choroidal neovascularization (CNV) associated with wet AMD does not display the hallmark vascular polyp-like dilations found in patients with PCV, distinguishing the two conditions. The primary environmental/behavioral risk factor for AMD is cigarette smoking, likely via an increase in systemic oxidative stress that promotes chronic inflammatory responses in the retina[23-25]. Chronic retinal inflammation is thought to accelerate degradation of the blood-retina barrier and trigger signaling cascades that increase pro-angiogenic gene expression associated with wet AMD[26]. The most prominent genetic risk factors for AMD include the nonsynonymous variant rs1061170 on chromosome 1q31, which encodes a Tyr402His substitution in complement factor H (CFH), and an ~5-kb high linkage-disequilibrium (LD) block spanning the age-related maculopathy susceptibility 2/high-temperature requirement A serine peptidase 1 (ARMS2-HTRA1) locus on chromosome 10q26[27-30]. Because the ARMS2-HTRA1 risk haplotype slightly favors progression to wet AMD, we focus this review on the interplay among ARMS2-HTRA1 genetic risk, cigarette smoking, and oxidation-induced inflammatory responses on wet AMD development. We conclude with a mechanistic model of wet AMD progression that we hope will stimulate future investigations on wet AMD and other neovascular diseases.
Comparison of risk factors and clinical features between AMD and related retinal dystrophies
| Disease | Risk factors | Clinical features | Ref. |
|---|---|---|---|
| Age-related macular degeneration (AMD) | Age > 55 years is the greatest risk factor
Genetic: strongly associated with variants in the CFH and ARMS2-HTRA1 loci; other variants also implicated Environmental: cigarette smoking, high body mass index, other oxidative stressors | Development of drusen followed by geographic atrophy (GA) and/or choroidal neovascularization (CNV) | [5,6] |
| Polypoidal Choroidal Vasculopathy (PCV) | Like AMD, age > 55 years is a risk factor
Genetic: associated with AMD risk variants in the ARMS2-HTRA1, CFH, Elastin and C2 loci. Notably, only rs10490924, an ARMS2/LOC38771 coding variant, displays a statistically significant difference between PCV and AMD risk Environmental: cigarette smoking, high body mass index, other oxidative stressors | Abnormal branching network of blood vessels displaying polyps or polypoidal dilations within the Bruch’s Membrane in addition to choroidal thickening | [7-10] |
| Doyne Honeycomb Retinal Dystrophy | Genetic: R345W variant in EFEMP1, which encodes Fibulin-3, an ECM protein | Radial/honeycomb pattern of drusen formation leading into geographic atrophy and choroidal neovascularization | [11-13] |
| Sorby’s Fundus Dystrophy | Genetic: loss of function missense and nonsense variants in TIMP3, an MMP regulator | Accumulation of drusen in the subretinal space preceding subretinal hemorrhaging; RPE detachment and atrophy; and possibly choroidal neovascularization | [14,15] |
| Stargardt Disease | Genetic: 95% of cases are attributed to variants in ABCA4, an ATP-binding cassette transporter protein. The remaining 5% of cases are associated with variants in STGF4, ELOVL4, and PRPH2 | Accumulation of A2E and lipofuscin in RPE cells, leading to dyschromatopsia and macular degeneration. Several variants are associated with “fovea-sparing” | [16,17] |
| Best Vitelliform Macular Dystrophy | Genetic: variants in BEST1, encoding an ion channel protein that regulates Ca2+ signaling | Lipofuscin accumulation within the RPE and macular vitelliform lesions, leading to cellular atrophy and sometimes choroidal neovascularization | [18,19] |
| Angioid (Knapp)
Streaks | Phenotype associated with Pseudoxanthoma Elasticum, Paget Disease, Ehlers-Danlos Syndrome, sickle cell hemoglobinopathies, and other diseases | Calcification of Bruch’s Membrane, leading to the development of linear breaks, lesions, and choroidal neovascularization | [20-22] |
Pathology of wet AMD
AMD leads to degeneration of the macula, the cone-rich central portion of the retina that is adapted for high-acuity daytime color vision [Figure 1][31]. It primarily affects the blood-retina barrier, consisting of the retinal pigment epithelium (RPE) and underlying Bruch’s Membrane (BM), and secondarily the photoreceptors [Figure 2A]. Proper phototransduction by photoreceptors is reliant on an intact and functional blood-retina barrier for uptake of essential nutrients from the choroidal vasculature, disposal of cellular waste to systemic circulation, and regeneration of the active chromophore 11-cis-retinal via the visual cycle, among other functions[32,33]. This delicate balance is disrupted in the AMD eye. The first clinical manifestations of disease are drusen, which appear on color fundus as yellowish focal deposits beneath the retina [Figure 2B and C]. They accumulate in the extracellular space between the RPE and BM and have recently been suggested to result from the oligomerization of lipids and proteins onto hydroxyapatite spherules[34]. Regardless of their origin, the size and number of drusen are used to determine an individual’s disease stage and assess the likelihood of progression to advanced AMD with severe central vision loss[35,36]. “Soft” drusen with poorly demarcated boundaries or diameter greater than 125 µm are particularly prognostic for severe AMD[37,38]. Unfortunately, drusen characteristics do not readily distinguish between individuals who will develop dry vs. wet AMD[38].
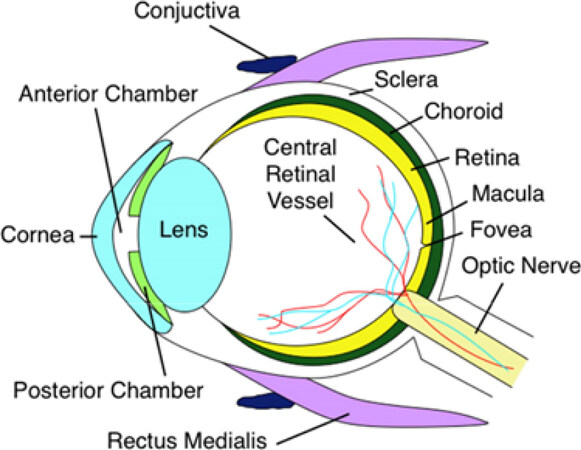
Figure 1. Anatomical cross section of the human eye. The macula is the central portion of the retina, responsible for high-acuity vision. Age-related macular degeneration progression leads to macular dystrophy, death of overlying photoreceptors, and eventually central vision loss
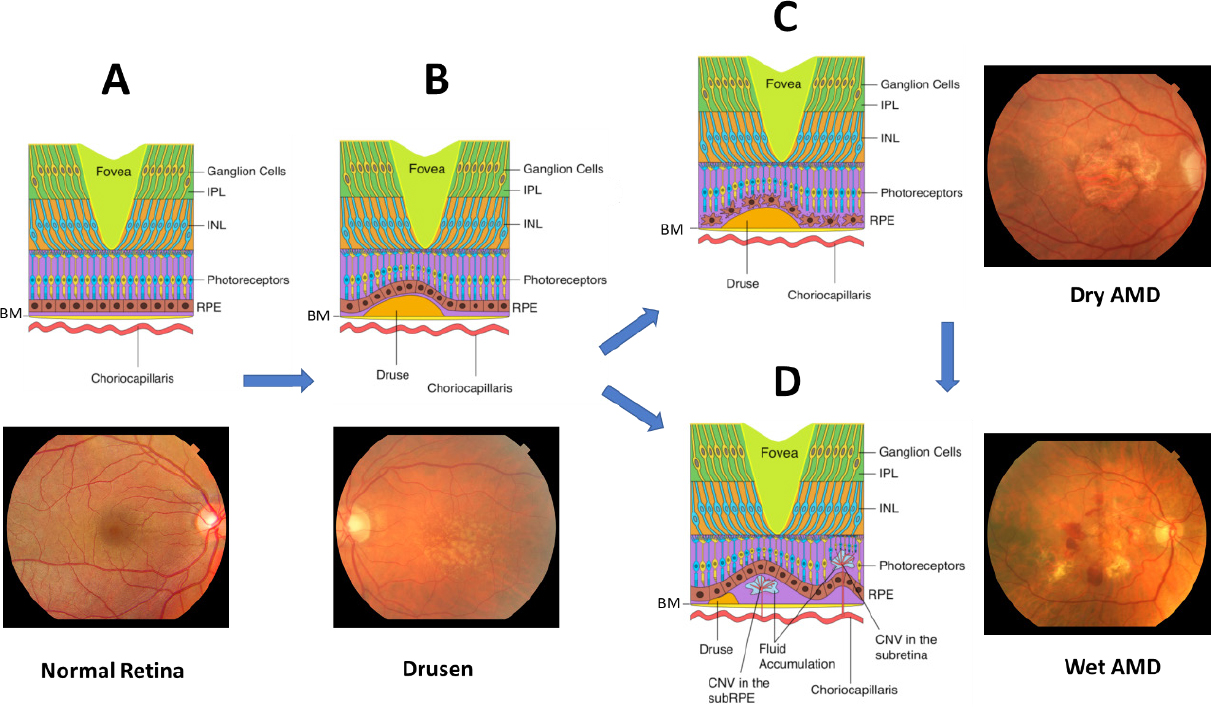
Figure 2. The clinical features of AMD progression. (A) Cross section and fundus photo of normal retina showing BM; choriocapillaris; IPL; INL; photoreceptors; and RPE; (B) drusen form underneath the RPE cell layer, preceding late-stage AMD; (C) dry AMD is characterized by extensive drusen, atrophy of RPE and photoreceptors, and deformed blood-retina barrier; (D) wet AMD is characterized by choroidal neovascularization across Bruch’s Membrane into the macula, leading to subretinal hemorrhage and possibly retinal detachment. AMD: age-related macular degeneration; BM: Bruch’s Membrane; IPL: inner plexiform layer; INL: inner nuclear layer; RPE: retinal pigment epithelium
In the early stages of wet AMD, inflammatory cells are observed in the subretinal space between the RPE and BM[39]. They disrupt the integrity of the blood-retina barrier through the release of inflammatory oxidants and pro-angiogenic cytokines and chemokines, including vascular endothelial growth factor (VEGF). VEGF stimulates endothelial cells to proliferate, migrate, and germinate to form new capillaries that cross the BM into the neural retina, a process referred to as CNV [Figure 2D][40,41]. VEGF mediates the expression of MCP-1, an angiogenic chemokine that recruits more macrophages to the retina[42,43], and acts as a specific endothelial cell mitogen to increase vascular permeability[44]. The structural integrity of the blood-retina barrier is further compromised by macrophage matrix metalloproteinases (MMP), which degrade the pentalaminar fibrous proteins of the BM[45-49]. As CNV expands within the subretinal space, the macula deforms and subretinal hemorrhaging allow cells and fluid in circulation to leak into the neural retina[50]. In its most advanced stages, CNV sites become fibrotic and form disciform scars, leading to necrosis of RPE and photoreceptors[33,50]. Photoreceptor cell death secondary to these processes is the immediate cause of vision loss for the patient.
Redox biochemistry and oxidative stress in the retina
Introduction to oxidation in the retina
Free reactive oxygen species (ROS) can react with biological macromolecules to form oxidation-modified, pro-inflammatory species[51,52]. Photoreceptor outer segments are rich in polyunsaturated fatty acids, which are easily oxidized by photosensitizers in the retina[53]. As these highly oxidized outer segments are shed, they are phagocytosed by the RPE through a tightly regulated sequence of steps that involves outer segment recognition and binding by the integrin αvβ5[54,55] and the scavenger receptor CD36[56], followed by activation of MERTK to trigger internalization[57]. Once inside the RPE, the outer segments are degraded along the phagolysosomal pathway into compounds that are either recycled back into the visual cycle or exocytosed from the RPE basolateral membrane into the choroid for clearance from systemic circulation[32]. The continuous shedding of oxidized outer segments is a major phagocytic challenge for the RPE that heightens intracellular oxidative stress and may contribute to age-related accumulation of oxidation-modified lipids and proteins in the retina, leading to disease[58,59].
The inflammatory response to oxidative stress
As people age, the permeability of Bruch’s Membrane decreases, thereby hindering the transport of exocytosed waste products from the RPE to the choroid[60]. These waste products accumulate in the sub-RPE space, prolonging their exposure to the oxidizing extracellular environment. Oxidation-specific epitopes, also referred to as Damage-Associated Molecular Patterns (DAMPs), on otherwise normal lipids and proteins, signal to the cell that the parent compound is damaged, flagging it for clearance by the immune system[61]. Many cytokines and chemokines are recruited during the inflammatory response, promoting angiogenesis and CNV [Table 2].
Inflammatory agents implicated in wet AMD progression
| Protein | Location | Function | Ref. |
|---|---|---|---|
| IL-6 | Soluble | Induces expression of angiogenic cytokines, including VEGF; regulates CNV progression | [62-64] |
| IL-8 | Soluble | Pro-angiogenic factor; strong chemotactic factor for neutrophils; elicits VEGF-A and VEGFR2 expression by endothelial cells | [65-67] |
| MCP-1/CCL2 | Soluble | Regulates migration and infiltration of monocytes and macrophages; participates in VEGF-induced angiogenesis; and contributes to the formation of sub-foveal choroidal neovascular membranes | [63,68-70] |
| CCR2 | Membrane | Promotes CNV formation by enhancing recruitment of myeloid cells | [68] |
| ICAM-1 | Membrane | Associates with inflammatory cells in subretinal neovascular lesions | [71] |
| VEGF | Soluble | Most prominent pro-angiogenic factor that stimulates CNV | [72] |
| IP-10 | Soluble | Associated with CNV | [73] |
| TGF-β | Soluble | Both promotes and inhibits CNV via diverse signaling pathways | [74,75] |
| IFN-γ | Soluble | Anti-angiogenic cytokine; induces expression of CFH and major histocompatibility complex-II in RPE cells | [76,77] |
| IGF-1 | Soluble | Pro-angiogenic cytokine; promotes CNV | [78,79] |
DAMPs are bound by a variety of Pattern Recognition Receptors (PRRs), including scavenger receptors, toll-like receptors, C-reactive protein, complement components, and IgM antibodies[80,81]. Interestingly, drusen have also been found to contain C-reactive protein, immunoglobulins, and many components of the complement pathway[82,83]. The presence of these complement proteins and oxidation products has been shown to result in changes to the cellular composition of the RPE and choroid. However, before these changes occur, cellular oxidative stress must elicit the secretion of pro-inflammatory cytokines and chemokines, overwhelming the cellular regulatory mechanisms. In the next section, we will review how this redox regulatory system becomes overburdened in the AMD eye.
Overview of redox biochemistry
Biochemical oxidation-reduction reactions are central components of many metabolic and signaling pathways[84]. As such, they are tightly regulated using a variety of enzymatic and non-enzymatic mechanisms, which control the balance of pro-oxidant and antioxidant species[52]. One of the most studied redox regulators is glutathione (GSH), a tripeptide of glutamate, cysteine, and glycine[85]. When exposed to oxidizing agents, glutathione’s thiol moiety is oxidized to form a disulfide bridge with another molecule of glutathione, forming glutathione disulfide (GSSG)[86]. GSSG can be reduced by glutathione reductase (GSH reductase) at the expenditure of one NADPH, reforming two GSH[86]. Together, the interconversion of GSH and GSSG forms a redox buffer system[87], analogous to the bicarbonate buffer system for pH.
The status of the redox buffer system can be used to assess the oxidative state of the intracellular environment. Studies have shown that the GSH:GSSG ratio remains within narrow ranges that are specific to particular intracellular compartments[88]. The GSH:GSSG ratio of the endoplasmic reticulum ranges from 1:1 to 1:3[89,90], whereas the GSH:GSSG ratio of the cytosol ranges from 30:1 to 100:1[90]. However, chronic overproduction of oxidizing agents can alter redox homeostasis and decrease the GSH:GSSG ratio, which has been correlated with aging and disease[91,92]. The greatest endogenous source of oxidizing agents is release of ROS from the mitochondrial matrix as byproducts of oxidative phosphorylation[93]. As electrons move along the electron transport chain, some electrons leak from Complexes I and III, forming superoxide radical (O2-)[94]. Superoxide radical is scavenged by superoxide dismutase (SOD) to form hydrogen peroxide (H2O2), which may be either reduced to water by glutathione peroxidase (GPx) at the expenditure of one glutathione or dismutated to water and molecular oxygen by catalase (CAT)[95]. However, excess leakage of O2- can overload these enzymatic mechanisms, resulting in increased ROS concentration in the cytosol and nucleus [Figure 3]. Excess ROS can lead to nuclear and mitochondrial DNA damage, autophagy decay, and apoptosis of photoreceptors and RPE via the MAPK/ERK 1/2 pathway[96-99]. In addition to an endogenous leakage of mitochondrial ROS, cigarette smoke, a prominent environmental/behavioral risk factor for AMD[100-103], contains an assortment of free radicals and ROS that disturb cellular redox[104].
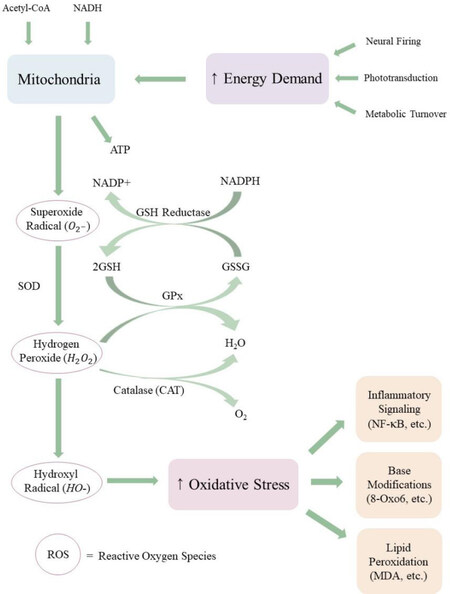
Figure 3. The flow of ROS in the retina. Mitochondria respond to high energy demand in the retina by synthesizing ATP, a by-product of which is the potent ROS superoxide radical. Superoxide radical is converted to less reactive species through a sequence of enzymatic and non-enzymatic pathways. If mitochondrial ROS release exceeds antioxidant scavenging capacity, ROS accumulate in the retina and can react with macromolecules, including DNA and unsaturated phospholipids. Elevated ROS and oxidation-modified by-products elicit chronic inflammation, contributing to retinal pathologies like AMD. ROS: reactive oxygen species; AMD: age-related macular degeneration
Oxidative stress in the retina
The retina is especially prone to increased endogenous ROS production due to its high energy demand, high dissolved oxygen concentration, and dependency on mitochondrial oxidative phosphorylation for proper function[105,106]. Photoreceptor mitochondria are predominantly clustered within the inner segment, whereas the outer segment is sparsely populated[107]. Unlike most neurons, photoreceptors are depolarized in their unstimulated state and are hyperpolarized upon stimulation with light, forming the dark current[108]. In the absence of light, Na+/K+ ATPases are actively maintaining a continual flux of Na+ into the cell, enabling continual release of the excitatory neurotransmitter glutamate into the photoreceptor-bipolar cell synaptic cleft[109]. This ATPase activity accounts for most retinal energy requirements[109,110]. The RPE is also densely populated with mitochondria, which are used to power phagocytosis of photoreceptor outer segments; metabolic exchange between the neural retina and the choroid; and maintenance of transepithelial potential[107,111-113].
ARMS2-HTRA1 genetic risk and HTRA1 activity in wet AMD
Introduction to ARMS2-HTRA1 in AMD: discovery and controversy
In addition to cigarette smoking and oxidative stress, many genetic variants have been linked to AMD pathogenesis. The first genome-wide association studies (GWAS) on AMD identified variants in the complement factor H (CFH) locus on chromosome 1q31 that strongly associate with progression to late-stage AMD[114-117]. Shortly thereafter, two studies simultaneously discovered another strong genetic risk locus on chromosome 10q26[118,119]. These risk variants span a ~5-kb region of high linkage disequilibrium (LD) that includes the promoter of HtrA serine peptidase 1 (HTRA1), a secreted serine protease that has been extensively studied for its role in extracellular matrix (ECM) remodeling[120-122], transforming growth factor β (TGF-β) signaling[123-127], and various cancers[128-130]. Preliminary evidence from luciferase reporter assays using HTRA1 promoter sequences[118] and RT-PCR of HTRA1 mRNA from blood lymphocytes[119] indicated that the AMD risk haplotype increases HTRA1 expression ~2-fold. These findings support a putative disease mechanism in which chronically elevated HTRA1 due to the risk haplotype alters ECM homeostasis along the blood-retina barrier, leading to AMD. However, the risk haplotype also extends into the coding sequence for age-related maculopathy susceptibility 2 (ARMS2), an 11-kDa protein whose structure, function, and localization have been disputed since its discovery. Initially thought to be a mitochondrial protein[131,132], ARMS2 is now known to be secreted from cells[133]via a Golgi-independent pathway[134] and may regulate complement activation[135]. However, ARMS2 seems to be an unlikely source for AMD pathogenicity due to (1) its weak expression in the retina[131,132,136,137] and (2) a recent expression quantitative trait loci (eQTL) analysis that found multiple AMD-associated eQTLs that affect HTRA1 expression, but no AMD-associated eQTLs that exclusively affect ARMS2 expression[138]. While it is our view that HTRA1 is the more plausible candidate for AMD pathogenicity, we emphasize the pressing need for future studies to carefully isolate the potential roles of ARMS2 and HTRA1 in AMD pathogenesis to pinpoint the causal agent. In this review, we will focus on the evidence for HTRA1’s role in AMD pathogenesis and its interplay with aging, oxidative stress, and other genetic risk factors that predispose development of wet AMD [Figure 4].
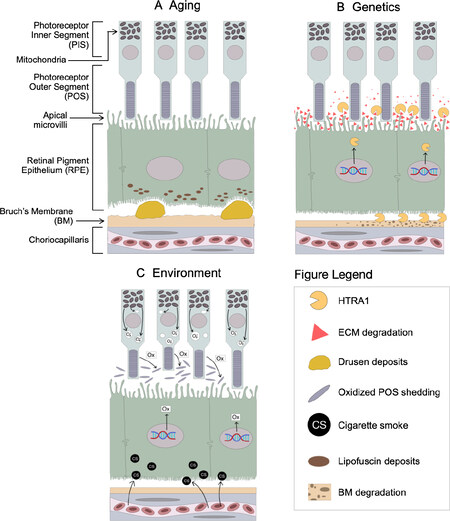
Figure 4. Retinal impact of wet AMD risk factors. (A) Depicts age-related lipofuscin accumulation and drusen formation; (B) the ARMS2-HTRA1 locus is a prominent genetic risk factor for AMD, resulting in excess HTRA1 expression and heightened degradation of RPE extracellular matrix and Bruch’s Membrane; (C) oxidative stress due to mitochondrial superoxide release and bloodborne cigarette smoke extracts alter transcriptional pathways that increase oxidative load in the retina. AMD: age-related macular degeneration; RPE: retinal pigment epithelium
Evidence for HTRA1’s role in wet AMD
HTRA1 is a 51-kDa secreted protein that belongs to the high temperature requirement A (HtrA) family of chymotrypsin-like serine proteases, which includes the paralogs HTRA2, HTRA3, and HTRA4[139,140]. Originally discovered in E. coli as an integral component of the heat shock response[141,142], HTRA1 is widely expressed in human tissue and has been implicated in many physiological processes, notably ECM remodeling[120-122] and TGF-β signaling[123-127]. Since the AMD risk haplotype does not extend into the HTRA1 coding region, researchers have long suspected that chronic excess HTRA1 expression mediates ARMS2-HTRA1 genetic risk by altering ECM physiology at the blood-retina barrier. These changes may result from either proteolytic degradation of Bruch’s Membrane or dysregulation of signaling pathways - especially TGF-β - that compromise blood-retina barrier integrity and/or promote CNV. It is important to note that these processes are not mutually exclusive and may have varying relevance to disease progression in specific AMD patients.
The preceding mechanisms share the common assumption that the ARMS2-HTRA1 risk haplotype increases retinal expression of HTRA1 in AMD patients, but this assertion remains unsettled due to conflicting results. Early studies of HTRA1 expression noted excess HTRA1 levels in patients harboring the AMD risk haplotype, but the sample sizes were generally small (< 10 subjects)[119,143-145]. In contrast, later studies with larger patient cohorts (> 30 subjects) oftentimes did not observe a significant difference in HTRA1 expression between patients harboring the protective and risk haplotypes[146-148]. However, these apparently contradictory findings may be resolved by impinging environmental factors, chiefly oxidative stress. Recent studies have found that oxidative stress induces HTRA1 expression[149-151]. Oka and colleagues were the first to show that oxidative stress elicits HTRA1 expression in the human RPE cell line ARPE-19 and murine embryonic fibroblasts (MEF)[150]. Excess HTRA1 induced cellular senescence by activating the p38 mitogen-activated protein kinase (MAPK) pathway, which was dependent on HTRA1 proteolytic activity. In our previous study, ARPE-19 cells challenged at their apical surface with oxidized low-density lipoprotein, which mimics the shedding of highly oxidized photoreceptor outer segments, had significantly increased HTRA1 levels but surprisingly had no change in VEGF levels 24 h after stimulation[151]. In contrast, when these cells were exposed to secretions from macrophages under oxidative stress, both HTRA1 and VEGF levels increased, likely via the Wnt signaling cascade. These results highlight the role of oxidative stress in exacerbating HTRA1 expression associated with the AMD risk haplotype and the role of pro-angiogenic factors secreted by macrophages in accelerating neovascularization in the wet AMD eye.
Assuming HTRA1 expression is increased by the ARMS2-HTRA1 risk haplotype and/or under heightened oxidative stress, chronic excess HTRA1 would be predicted to compromise the structural integrity of the blood-retina barrier. HTRA1 is known to cleave many transmembrane and secreted proteins in the ECM, including critical components of Bruch’s Membrane. Major structural ECM proteins known to be substrates of HTRA1 proteolysis include decorin[152,153], EGF-containing fibulin-like extracellular matrix protein 1 (EFEMP1)[154], fibronectin[122,153,155], fibromodulin[120,152], nidogen-1[122], nidogen-2[122], thrombospondin-1[143,151], type II collagen[152], and vitronectin[120]. Gradual degradation of Bruch’s Membrane opens pathways for vascular penetrance from the choroid into the neural retina, heightening the risk of wet AMD development. An underappreciated aspect of this process is that the ECM fragment by-products of HTRA1 proteolytic digestion may themselves be pro-angiogenic, exacerbating risk for wet AMD beyond the breakdown of physical separation between the choroid and neural retina. For example, it was recently shown that the N-terminal fragment of thrombospondin-1 produced by HTRA1 digestion promotes endothelial tube formation, whereas the parent thrombospondin-1 is anti-angiogenic[143]. Presumably, the parallel processes of structural proteolysis and ECM fragment signaling always have the potential to mediate rapid breakdown of the blood-retina barrier. Future studies are needed to elucidate the regulatory mechanisms that keep these potentially deleterious processes within healthy limits and to identify the age-related or AMD risk factor-specific changes that disrupt these safeguards, leading to pathology.
HTRA1 also regulates TGF-β signaling, which serves diverse functions in adults and developing embryos[156]. The pathway is triggered by ligand binding to TGF-β receptors, followed by phosphorylation of SMAD proteins, which then form complexes that translocate into the nucleus and act as transcription factors[156]. HTRA1 has been implicated in cleaving multiple members of the TGF-β protein family, including mature TGF-β[125] (although this is disputed[123,126]), proTGF-β1[126], Type II and Type III TGF-β receptors[123], and latent TGF-β binding protein 1 (LTBP1)[127]. Regardless of its mechanism of action, HTRA1 proteolytic activity decreases TGF-β signaling, leading researchers to explore the consequences of downregulated TGF-β on AMD pathogenesis. Zhang and colleagues suggested that excess HTRA1 promotes angiogenesis via growth differentiation factor 6 (GDF6), a member of the TGF-β family[145]. In patients harboring the ARMS2-HTRA1 risk variant rs10490924, they noted increased HTRA1 and decreased GDF6. Conversely, in HTRA1-knockout mice, they observed increased GDF6 and decreased VEGF levels, suggesting that the HTRA1-null allele is protective against VEGF-mediated retinal neovascularization. Additionally, a study of Type II TGF-β receptor knockout mice (TGFBR2-/-) at 3-weeks and 6-months exhibited clear signs of wet AMD, including deformed RPE, mononuclear cells in the subretinal space, and degraded photoreceptor outer segments[157]. Critically, VEGF was significantly increased in these mice, suggesting that insufficient TGF-β signaling can induce CNV. Schlecht and colleagues proposed that VEGF and TGF-β serve antagonistic roles in maintaining endothelial homeostasis[157]. Since HTRA1-mediated cleavage of either TGF-β or its receptors decreases overall TGF-β signaling, it is plausible that chronic excess HTRA1 induces CNV because there is insufficient TGF-β to counteract VEGF activity. However, more research is needed to identify the precise mechanism by which decreased TGF-β activity accelerates AMD pathogenesis.
Genetic insights suggest a new model for wet AMD progression
For years, research on AMD pathogenesis has largely focused on the two most prominent genetic risk loci as unveiled through GWAS: ARMS2-HTRA1 and CFH. However, critical perspectives can be gleaned by assessing genetic risk factors that predispose development to wet AMD vs. dry AMD. In the largest GWAS on AMD to date, Fritsche and colleagues found 52 variants distributed across 34 loci that are associated with progression to late-stage AMD, either dry or wet[158]. Interestingly, only four loci harbored variants that significantly predispose development to the wet subtype: ARMS2-HTRA1 on chromosome 10, CETP on chromosome 16, MMP9 on chromosome 20, and SYN3-TIMP3 on chromosome 22 (mutations in this locus are also associated with Sorby’s Fundus Dystrophy, an AMD-like phenotype). While these four loci predispose development to wet AMD over dry AMD, the MMP9 locus is unique in that its risk variant is only associated with wet AMD, not dry AMD. This region encodes matrix metalloproteinase 9 (MMP9), which, like HTRA1, is a critical regulator of ECM remodeling. MMP9 is known to participate in positive feedback with VEGF; when MMP9 levels increase, so do VEGF levels, and vice versa[159]. Even more striking, MMP9 expression in cultured RPE cells is elicited by exposure to fibronectin fragments, which notably are the byproduct of HTRA1-mediated degradation of fibronectin[160]. Chronically elevated fibronectin fragment levels along the blood-retina barrier due to excess HTRA1 expression would also be expected to increase MMP9 expression. Much like the pro-angiogenic N-terminal fragment of thrombospondin-1, excess MMP9 would not only accelerate Bruch’s Membrane breakdown, but also stimulate VEGF expression, thereby triggering neovascularization. Over time, elevated expression of HTRA1 in dry AMD patients harboring the ARMS2-HTRA1 risk haplotype may elicit sufficient MMP9 and, subsequently, VEGF to trigger the transition to wet AMD. While it remains unclear why only some dry AMD patients develop wet AMD, these findings suggest that the HTRA1-MMP9 axis may be the critical intermediate between ARMS2-HTRA1 genetic risk and wet AMD [Figure 5].
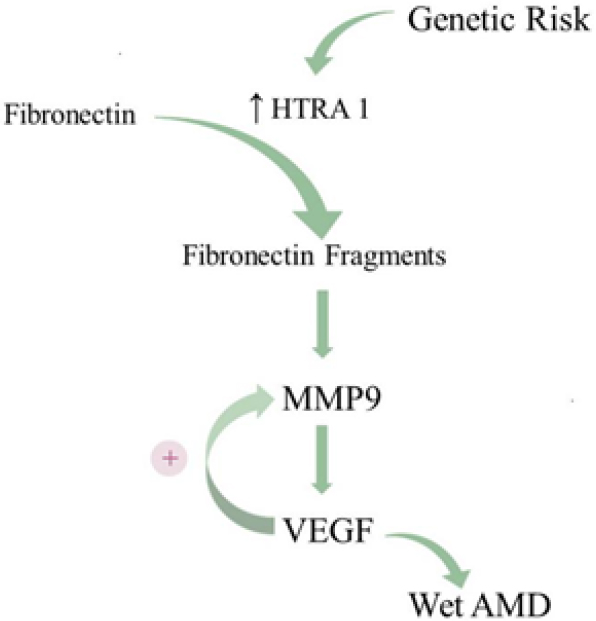
Figure 5. The proposed HTRA1-MMP9 axis in wet AMD. The ARMS2-HTRA1 risk haplotype increases expression of HTRA1, heightening the degradation of ECM constituents. HTRA1-mediated cleavage of the ECM protein fibronectin generates fibronectin fragments, which stimulate MMP9 expression. Positive feedback between MMP9 and VEGF promotes CNV and leads to wet AMD. AMD: age-related macular degeneration; ECM: extracellular membrane; VEGF: vascular endothelial growth factor; CNV: choroidal neovascularization
Conclusion: new avenues for investigation and therapeutic intervention
Despite major advancements in our understanding of the risk factors for AMD through GWAS, eQTL, and longitudinal case studies, there is a dearth of effective treatment options for patients living with AMD. Currently, the only robustly effective treatment for wet AMD is anti-VEGF therapy (e.g., ranibizumab, bevacizumab). While anti-VEGF therapy has undoubtedly revolutionized clinical management of wet AMD, this approach targets a late downstream effector of CNV, requires repeated monthly injections to sustain therapeutic effect, and is ineffective in a substantial portion of wet AMD patients[161,162]. Above all, anti-VEGF offers no benefit to people living with dry AMD, who constitute roughly 90% of all AMD cases. The lack of novel and effective alternative approaches to treat both wet and dry AMD is a pressing unmet medical need facing ophthalmologists worldwide. Our analysis suggests that HTRA1 and MMP9 are strong candidate therapeutic targets that warrant further investigation. Because HTRA1 is considerably upstream of VEGF and is associated with both wet and dry AMD, anti-HTRA1 therapy holds promise of slowing or halting the progression of both wet and dry AMD. Our lab has uncovered encouraging preliminary evidence that anti-HTRA1 single-chain variable fragment (scFv) can significantly reduce the size of neovascular lesions in a laser-induced mouse model of wet AMD[151]. Further support for this approach comes from a recent report by Lill and colleagues, who independently generated an anti-HTRA1 Fab and demonstrated that Dickkopf-related protein 3 (DKK3) levels in the aqueous humor of patients with dry AMD could serve as a biomarker for HTRA1 proteolytic inhibition by their anti-HTRA1 Fab[163]. Despite these early successes, HTRA1’s position upstream of VEGF implies that inhibiting its proteolytic activity will affect a greater number of physiological pathways in the retina, increasing the likelihood of deleterious side effects. For this reason, MMP9 may be a viable alternative or combinatorial therapeutic target in patients with wet AMD who do not respond adequately to anti-VEGF alone. Regardless of which therapeutic approach is ultimately most successful, these investigations will yield valuable insights into the pathways leading from genetic risk to AMD pathology and potentially uncover new ways of conceptualizing and treating other neovascular diseases.
Declarations
Authors’ contributionsConceptualized and prepared the original draft: Lu ZG, May A, Dinh B, Lin V, Shaw PX
Drafted and revised figures: Su F, Tran C, Borooah S, Adivikolanu H
Reviewed and edited the manuscript: May A, Dinh B, Che B, Wang ZH, Ehlen R, Shaw DH
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by National Institutes of Health Grants (RO1EY023693) (to Shaw PX) and (P30EY022589).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration--emerging pathogenetic and therapeutic concepts. Ann Med 2006;38:450-71.
2. Friedman DS, O’Colmain BJ, Muñoz B, et al; Eye Diseases Prevalence Research Group. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol 2004;122:564-72.
4. Prokofyeva E, Zrenner E. Epidemiology of major eye diseases leading to blindness in Europe: a literature review. Ophthalmic Res 2012;47:171-88.
5. Ding X, Patel M, Chan CC. Molecular pathology of age-related macular degeneration. Prog Retin Eye Res 2009;28:1-18.
6. Al-Zamil WM, Yassin SA. Recent developments in age-related macular degeneration: a review. Clin Interv Aging 2017;12:1313-30.
7. Chen H, Liu K, Chen LJ, Hou P, Chen W, Pang CP. Genetic associations in polypoidal choroidal vasculopathy: a systematic review and meta-analysis. Mol Vis 2012;18:816-29.
8. Wong CW, Yanagi Y, Lee WK, et al. Age-related macular degeneration and polypoidal choroidal vasculopathy in Asians. Prog Retin Eye Res 2016;53:107-39.
9. Cheung CMG, Lai TYY, Ruamviboonsuk P, et al. Polypoidal Choroidal Vasculopathy: Definition, Pathogenesis, Diagnosis, and Management. Ophthalmology 2018;125:708-24.
10. Kondo N, Honda S, Ishibashi K, Tsukahara Y, Negi A. Elastin gene polymorphisms in neovascular age-related macular degeneration and polypoidal choroidal vasculopathy. Invest Ophthalmol Vis Sci 2008;49:1101-5.
11. Stone EM, Lotery AJ, Munier FL, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet 1999;22:199-202.
12. Marmorstein L. Association of EFEMP1 with malattia leventinese and age-related macular degeneration: a mini-review. Ophthalmic Genet 2004;25:219-26.
13. Livingstone I, Uversky VN, Furniss D, Wiberg A. The Pathophysiological Significance of Fibulin-3. Biomolecules 2020;10:1294.
14. Gliem M, Müller PL, Mangold E, et al. Sorsby Fundus Dystrophy: Novel Mutations, Novel Phenotypic Characteristics, and Treatment Outcomes. Invest Ophthalmol Vis Sci 2015;56:2664-76.
15. Qi JH, Bell B, Singh R, et al. Sorsby Fundus Dystrophy Mutation in Tissue Inhibitor of Metalloproteinase 3 (TIMP3) promotes Choroidal Neovascularization via a Fibroblast Growth Factor-dependent Mechanism. Sci Rep 2019;9:17429.
16. Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol 2017;101:25-30.
17. Tsang SH, Sharma T. Stargardt Disease. In: Tsang SH, Sharma T, editors. Atlas of Inherited Retinal Diseases. Cham: Springer International Publishing; 2018. pp. 139-51.
18. Guziewicz KE, Sinha D, Gómez NM, et al. Bestrophinopathy: An RPE-photoreceptor interface disease. Prog Retin Eye Res 2017;58:70-88.
19. Johnson AA, Guziewicz KE, Lee CJ, et al. Bestrophin 1 and retinal disease. Prog Retin Eye Res 2017;58:45-69.
20. Al-Rashaed S, Arevalo JF. Long-term follow-up of choroidal neovascularization secondary to angioid streaks: case series and literature review. Clin Ophthalmol 2012;6:1029-34.
21. Martinez-Serrano MG, Rodriguez-Reyes A, Guerrero-Naranjo JL, et al. Long-term follow-up of patients with choroidal neovascularization due to angioid streaks. Clin Ophthalmol 2017;11:23-30.
22. Chatziralli I, Saitakis G, Dimitriou E, et al. ANGIOID STREAKS: A Comprehensive Review From Pathophysiology to Treatment. Retina 2019;39:1-11.
23. Shaw PX, Stiles T, Douglas C, et al. Oxidative stress, innate immunity, and age-related macular degeneration. AIMS Mol Sci 2016;3:196-221.
24. Kauppinen A, Niskanen H, Suuronen T, Kinnunen K, Salminen A, Kaarniranta K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells--implications for age-related macular degeneration (AMD). Immunol Lett 2012;147:29-33.
25. Hollyfield JG, Bonilha VL, Rayborn ME, et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med 2008;14:194-8.
26. Datta S, Cano M, Ebrahimi K, Wang L, Handa JT. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog Retin Eye Res 2017;60:201-18.
27. Sobrin L, Reynolds R, Yu Y, et al. ARMS2/HTRA1 locus can confer differential susceptibility to the advanced subtypes of age-related macular degeneration. Am J Ophthalmol 2011;151:345-52.e3.
28. Askari M, Nikpoor AR, Gorjipour F, et al. Association of Htra1 gene polymorphisms with the risk of developing AMD in Iranian population. Rep Biochem Mol Biol 2015;4:43-9.
29. Andreoli MT, Morrison MA, Kim BJ, et al. Comprehensive analysis of complement factor H and LOC387715/ARMS2/HTRA1 variants with respect to phenotype in advanced age-related macular degeneration. Am J Ophthalmol 2009;148:869-74.
30. Grassmann F, Heid IM, Weber BH; International AMD Genomics Consortium (IAMDGC). Recombinant Haplotypes Narrow the ARMS2/HTRA1 Association Signal for Age-Related Macular Degeneration. Genetics 2017;205:919-24.
31. Provis JM, Penfold PL, Cornish EE, Sandercoe TM, Madigan MC. Anatomy and development of the macula: specialisation and the vulnerability to macular degeneration. Clin Exp Optom 2005;88:269-81.
33. Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol Aspects Med 2012;33:295-317.
34. Thompson RB, Reffatto V, Bundy JG, et al. Identification of hydroxyapatite spherules provides new insight into subretinal pigment epithelial deposit formation in the aging eye. Proc Natl Acad Sci U S A 2015;112:1565-70.
35. Klein R, Meuer SM, Myers CE, et al. Harmonizing the classification of age-related macular degeneration in the three-continent AMD consortium. Ophthalmic Epidemiol 2014;21:14-23.
36. Ferris III FL, Wilkinson C, Bird A, et al. Clinical classification of age-related macular degeneration. Ophthalmology 2013;120:844-51.
37. Johnson PT, Lewis GP, Talaga KC, et al. Drusen-associated degeneration in the retina. Invest Ophthalmol Vis Sci 2003;44:4481-8.
38. Khan KN, Mahroo OA, Khan RS, et al. Differentiating drusen: Drusen and drusen-like appearances associated with ageing, age-related macular degeneration, inherited eye disease and other pathological processes. Prog Retin Eye Res 2016;53:70-106.
39. Penfold PL, Madigan MC, Gillies MC, Provis JM. Immunological and aetiological aspects of macular degeneration. Prog Retin Eye Res 2001;20:385-414.
40. Kwak N, Okamoto N, Wood JM, Campochiaro PA. VEGF is major stimulator in model of choroidal neovascularization. Invest Ophthalmol Vis Sci 2000;41:3158-64.
41. Ishibashi T, Hata Y, Yoshikawa H, Nakagawa K, Sueishi K, Inomata H. Expression of vascular endothelial growth factor in experimental choroidal neovascularization. Graefes Arch Clin Exp Ophthalmol 1997;235:159-67.
42. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 2009;29:313-26.
43. Hong KH, Ryu J, Han KH. Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood 2005;105:1405-7.
44. Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 2005;437:497-504.
45. Nita M, Strzałka-Mrozik B, Grzybowski A, Mazurek U, Romaniuk W. Age-related macular degeneration and changes in the extracellular matrix. Med Sci Monit 2014;20:1003-16.
46. Singh M, Tyagi SC. Metalloproteinases as mediators of inflammation and the eyes: molecular genetic underpinnings governing ocular pathophysiology. Int J Ophthalmol 2017;10:1308-18.
47. Pufe T, Harde V, Petersen W, Goldring MB, Tillmann B, Mentlein R. Vascular endothelial growth factor (VEGF) induces matrix metalloproteinase expression in immortalized chondrocytes. J Pathol 2004;202:367-74.
48. Huang WC, Sala-Newby GB, Susana A, Johnson JL, Newby AC. Classical macrophage activation up-regulates several matrix metalloproteinases through mitogen activated protein kinases and nuclear factor-κB. PLoS One 2012;7:e42507.
49. Bandyopadhyay M, Rohrer B. Matrix metalloproteinase activity creates pro-angiogenic environment in primary human retinal pigment epithelial cells exposed to complement. Invest Ophthalmol Vis Sci 2012;53:1953-61.
50. Mitchell P, Liew G, Gopinath B, Wong TY. Age-related macular degeneration. The Lancet 2018;392:1147-59.
51. Terman A, Brunk UT. Oxidative stress, accumulation of biological ‘garbage’, and aging. Antioxid Redox Signal 2006;8:197-204.
52. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007;39:44-84.
53. Léveillard T, Sahel JA. Metabolic and redox signaling in the retina. Cell Mol Life Sci 2017;74:3649-65.
54. Miceli MV, Newsome DA, Tate DJ Jr. Vitronectin is responsible for serum-stimulated uptake of rod outer segments by cultured retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 1997;38:1588-97.
55. Finnemann SC, Bonilha VL, Marmorstein AD, Rodriguez-Boulan E. Phagocytosis of rod outer segments by retinal pigment epithelial cells requires alpha(v)beta5 integrin for binding but not for internalization. Proc Natl Acad Sci U S A 1997;94:12932-7.
56. Ryeom SW, Sparrow JR, Silverstein RL. CD36 participates in the phagocytosis of rod outer segments by retinal pigment epithelium. J Cell Sci 1996;109:387-95.
57. Feng W, Yasumura D, Matthes MT, LaVail MM, Vollrath D. Mertk triggers uptake of photoreceptor outer segments during phagocytosis by cultured retinal pigment epithelial cells. J Biol Chem 2002;277:17016-22.
58. Vives-Bauza C, Anand M, Shiraz AK, et al. The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J Biol Chem 2008;283:24770-80.
59. Sparrow JR, Boulton M. RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res 2005;80:595-606.
60. Moore DJ, Clover GM. The effect of age on the macromolecular permeability of human Bruch’s membrane. Invest Ophthalmol Vis Sci 2001;42:2970-5.
61. Miller YI, Choi SH, Wiesner P, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res 2011;108:235-48.
62. Holzinger C, Weissinger E, Zuckermann A, et al. Effects of interleukin-1, -2, -4, -6, interferon-gamma and granulocyte/macrophage colony stimulating factor on human vascular endothelial cells. Immunol Lett 1993;35:109-17.
63. Jonas JB, Tao Y, Neumaier M, Findeisen P. Cytokine concentration in aqueous humour of eyes with exudative age-related macular degeneration. Acta Ophthalmol 2012;90:e381-8.
64. Rezar-Dreindl S, Sacu S, Eibenberger K, et al. The Intraocular Cytokine Profile and Therapeutic Response in Persistent Neovascular Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci 2016;57:4144-50.
65. Henkels KM, Frondorf K, Gonzalez-Mejia ME, Doseff AL, Gomez-Cambronero J. IL-8-induced neutrophil chemotaxis is mediated by Janus kinase 3 (JAK3). FEBS Lett 2011;585:159-66.
66. Lechner J, Chen M, Hogg RE, et al. Peripheral blood mononuclear cells from neovascular age-related macular degeneration patients produce higher levels of chemokines CCL2 (MCP-1) and CXCL8 (IL-8). J Neuroinflammation 2017;14:42.
67. Martin D, Galisteo R, Gutkind JS. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem 2009;284:6038-42.
68. Robbie SJ, Georgiadis A, Barker SE, et al. Enhanced Ccl2-Ccr2 signaling drives more severe choroidal neovascularization with aging. Neurobiol Aging 2016;40:110-9.
69. Dong A, Xie B, Shen J, et al. Oxidative stress promotes ocular neovascularization. J Cell Physiol 2009;219:544-52.
70. Yamada M, Kim S, Egashira K, et al. Molecular mechanism and role of endothelial monocyte chemoattractant protein-1 induction by vascular endothelial growth factor. Arterioscler Thromb Vasc Biol 2003;23:1996-2001.
71. Heidenkummer HP, Kampik A. Chirurgische Extraktion subretinaler Pseudotumoren bei altersbezogener Makuladegeneration (AMD). Klinische, morphologische und immunohistochemische Ergebnisse [Surgical extraction of subretinal pseudotumors in age related macular degeneration. Clinical, morphologic and immunohistochemical results]. Ophthalmologe 1995;92:631-9.
72. Agawa T, Usui Y, Wakabayashi Y, et al. Profile of intraocular immune mediators in patients with age-related macular degeneration and the effect of intravitreal bevacizumab injection. Retina 2014;34:1811-8.
73. Sakurada Y, Nakamura Y, Yoneyama S, et al. Aqueous humor cytokine levels in patients with polypoidal choroidal vasculopathy and neovascular age-related macular degeneration. Ophthalmic Res 2015;53:2-7.
74. Bai Y, Liang S, Yu W, et al. Semaphorin 3A blocks the formation of pathologic choroidal neovascularization induced by transforming growth factor beta. Mol Vis 2014;20:1258-70.
75. Zarranz-Ventura J, Fernández-Robredo P, Recalde S, et al. Transforming growth factor-beta inhibition reduces progression of early choroidal neovascularization lesions in rats: P17 and P144 peptides. PLoS One 2013;8:e65434.
76. Gabrielian K, Osusky R, Sippy BD, Ryan SJ, Hinton DR. Effect of TGF-beta on interferon-gamma-induced HLA-DR expression in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 1994;35:4253-9.
77. Wu Z, Lauer TW, Sick A, Hackett SF, Campochiaro PA. Oxidative stress modulates complement factor H expression in retinal pigmented epithelial cells by acetylation of FOXO3. J Biol Chem 2007;282:22414-25.
78. Castellino N, Longo A, Avitabile T, et al. Circulating insulin-like growth factor-1: a new clue in the pathogenesis of age-related macular degeneration. Aging (Albany NY) 2018;10:4241-7.
79. Grant MB, Mames RN, Fitzgerald C, Ellis EA, Aboufriekha M, Guy J. Insulin-like growth factor I acts as an angiogenic agent in rabbit cornea and retina: comparative studies with basic fibroblast growth factor. Diabetologia 1993;36:282-91.
80. Binder CJ, Chang MK, Shaw PX, et al. Innate and acquired immunity in atherogenesis. Nat Med 2002;8:1218-26.
81. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol 2016;16:485-97.
82. Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J 2000;14:835-46.
83. Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res 2001;20:705-32.
84. Yang Y, Sauve AA. NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta 2016;1864:1787-800.
85. Masella R, Di Benedetto R, Varì R, Filesi C, Giovannini C. Novel mechanisms of natural antioxidant compounds in biological systems: involvement of glutathione and glutathione-related enzymes. J Nutr Biochem 2005;16:577-86.
87. Jones DP, Carlson JL, Mody VC, Cai J, Lynn MJ, Sternberg P. Redox state of glutathione in human plasma. Free Radic Biol Med 2000;28:625-35.
88. Kaludercic N, Deshwal S, Di Lisa F. Reactive oxygen species and redox compartmentalization. Front Physiol 2014;5:285.
89. Bass R, Ruddock LW, Klappa P, Freedman RB. A major fraction of endoplasmic reticulum-located glutathione is present as mixed disulfides with protein. J Biol Chem 2004;279:5257-62.
90. Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992;257:1496-502.
91. Jones DP. Extracellular redox state: refining the definition of oxidative stress in aging. Rejuvenation Res 2006;9:169-81.
92. Banerjee R. Redox outside the box: linking extracellular redox remodeling with intracellular redox metabolism. J Biol Chem 2012;287:4397-402.
93. Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med 2000;29:222-30.
95. Wang Y, Branicky R, Noë A, Hekimi S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol 2018;217:1915-28.
96. Marchi S, Giorgi C, Suski JM, et al. Mitochondria-ros crosstalk in the control of cell death and aging. J Signal Transduct 2012;2012:329635.
97. Ivanova D, Zhelev Z, Aoki I, Bakalova R, Higashi T. Overproduction of reactive oxygen species - obligatory or not for induction of apoptosis by anticancer drugs. Chin J Cancer Res 2016;28:383-96.
98. Rohowetz LJ, Kraus JG, Koulen P. Reactive Oxygen Species-Mediated Damage of Retinal Neurons: Drug Development Targets for Therapies of Chronic Neurodegeneration of the Retina. Int J Mol Sci 2018;19:3362.
99. Masuda T, Shimazawa M, Hara H. Retinal Diseases Associated with Oxidative Stress and the Effects of a Free Radical Scavenger (Edaravone). Oxid Med Cell Longev 2017;2017:9208489.
100. Chakravarthy U, Wong TY, Fletcher A, et al. Clinical risk factors for age-related macular degeneration: a systematic review and meta-analysis. BMC Ophthalmol 2010;10:31.
101. Clemons TE, Milton RC, Klein R, Seddon JM, Ferris FL 3rd; Age-Related Eye Disease Study Research Group. Risk factors for the incidence of Advanced Age-Related Macular Degeneration in the Age-Related Eye Disease Study (AREDS) AREDS report no. 19. Ophthalmology 2005;112:533-9.
102. Smith W, Assink J, Klein R, et al. Risk factors for age-related macular degeneration. Ophthalmology 2001;108:697-704.
103. Seddon JM, George S, Rosner B. Cigarette smoking, fish consumption, omega-3 fatty acid intake, and associations with age-related macular degeneration: the US Twin Study of Age-Related Macular Degeneration. Arch Ophthalmol 2006;124:995-1001.
104. Zhao J, Hopke PK. Concentration of Reactive Oxygen Species (ROS) in Mainstream and Sidestream Cigarette Smoke. Aerosol Science and Technology 2012;46:191-7.
105. Niven JE, Laughlin SB. Energy limitation as a selective pressure on the evolution of sensory systems. J Exp Biol 2008;211:1792-804.
107. Andrews RM, Griffiths PG, Johnson MA, Turnbull DM. Histochemical localisation of mitochondrial enzyme activity in human optic nerve and retina. Br J Ophthalmol 1999;83:231-5.
108. Hagins W, Penn R, Yoshikami S. Dark Current and Photocurrent in Retinal Rods. Biophys J 1970;10:380-412.
109. Ames A, Li Y, Heher E, Kimble C. Energy metabolism of rabbit retina as related to function: high cost of Na+ transport. J Neurosci 1992;12:840-53.
110. Okawa H, Sampath AP, Laughlin SB, Fain GL. ATP consumption by mammalian rod photoreceptors in darkness and in light. Curr Biol 2008;18:1917-21.
111. Caldwell RB, Slapnick SM. Increased cytochrome oxidase activity in the diabetic rat retinal pigment epithelium. Invest Ophthalmol Vis Sci 1989;30:591-9.
112. Kevany BM, Palczewski K. Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda) 2010;25:8-15.
114. Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005;308:385-9.
115. Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science 2005;308:421-4.
116. Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005;308:419-21.
117. Zareparsi S, Branham KE, Li M, et al. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. Am J Hum Genet 2005;77:149-53.
118. Dewan A, Liu M, Hartman S, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science 2006;314:989-92.
119. Yang Z, Camp NJ, Sun H, et al. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science 2006;314:992-3.
120. An E, Sen S, Park SK, Gordish-Dressman H, Hathout Y. Identification of novel substrates for the serine protease HTRA1 in the human RPE secretome. Invest Ophthalmol Vis Sci 2010;51:3379-86.
121. Tiaden AN, Breiden M, Mirsaidi A, et al. Human serine protease HTRA1 positively regulates osteogenesis of human bone marrow-derived mesenchymal stem cells and mineralization of differentiating bone-forming cells through the modulation of extracellular matrix protein. Stem Cells 2012;30:2271-82.
122. Vierkotten S, Muether PS, Fauser S. Overexpression of HTRA1 leads to ultrastructural changes in the elastic layer of Bruch’s membrane via cleavage of extracellular matrix components. PLoS One 2011;6:e22959.
123. Graham JR, Chamberland A, Lin Q, et al. Serine protease HTRA1 antagonizes transforming growth factor-β signaling by cleaving its receptors and loss of HTRA1 in vivo enhances bone formation. PLoS One 2013;8:e74094.
124. Oka C, Tsujimoto R, Kajikawa M, et al. HtrA1 serine protease inhibits signaling mediated by Tgfbeta family proteins. Development 2004;131:1041-53.
125. Launay S, Maubert E, Lebeurrier N, et al. HtrA1-dependent proteolysis of TGF-beta controls both neuronal maturation and developmental survival. Cell Death Differ 2008;15:1408-16.
126. Shiga A, Nozaki H, Yokoseki A, et al. Cerebral small-vessel disease protein HTRA1 controls the amount of TGF-β1 via cleavage of proTGF-β1. Hum Mol Genet 2011;20:1800-10.
127. Beaufort N, Scharrer E, Kremmer E, et al. Cerebral small vessel disease-related protease HtrA1 processes latent TGF-β binding protein 1 and facilitates TGF-β signaling. Proc Natl Acad Sci U S A 2014;111:16496-501.
128. Baldi A, De Luca A, Morini M, et al. The HtrA1 serine protease is down-regulated during human melanoma progression and represses growth of metastatic melanoma cells. Oncogene 2002;21:6684-8.
129. Chien J, Campioni M, Shridhar V, Baldi A. HtrA serine proteases as potential therapeutic targets in cancer. Curr Cancer Drug Targets 2009;9:451-68.
130. Altobelli E, Angeletti PM, Morroni M, Profeta VF. HtrA1 as a promising tissue marker in cancer: a meta-analysis. BMC Cancer 2018;18:143.
131. Kanda A, Chen W, Othman M, et al. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proc Natl Acad Sci U S A 2007;104:16227-32.
132. Fritsche LG, Loenhardt T, Janssen A, et al. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet 2008;40:892-6.
133. Kortvely E, Hauck SM, Duetsch G, et al. ARMS2 is a constituent of the extracellular matrix providing a link between familial and sporadic age-related macular degenerations. Invest Ophthalmol Vis Sci 2010;51:79-88.
134. Kortvely E, Hauck SM, Behler J, Ho N, Ueffing M. The unconventional secretion of ARMS2. Hum Mol Genet 2016;25:3143-51.
135. Micklisch S, Lin Y, Jacob S, et al. Age-related macular degeneration associated polymorphism rs10490924 in ARMS2 results in deficiency of a complement activator. J Neuroinflammation 2017;14:4.
136. Jakobsdottir J, Conley YP, Weeks DE, Mah TS, Ferrell RE, Gorin MB. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet 2005;77:389-407.
137. Wang G, Spencer KL, Scott WK, et al. Analysis of the indel at the ARMS2 3’UTR in age-related macular degeneration. Hum Genet 2010;127:595-602.
138. Liao SM, Zheng W, Zhu J, et al. Specific correlation between the major chromosome 10q26 haplotype conferring risk for age-related macular degeneration and the expression of HTRA1. Mol Vis 2017;23:318-33.
140. Clausen T, Kaiser M, Huber R, Ehrmann M. HTRA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol 2011;12:152-62.
141. Zumbrunn J, Trueb B. Primary structure of a putative serine protease specific for IGF-binding proteins. FEBS Lett 1996;398:187-92.
142. Hu SI, Carozza M, Klein M, Nantermet P, Luk D, Crowl RM. Human HtrA, an evolutionarily conserved serine protease identified as a differentially expressed gene product in osteoarthritic cartilage. J Biol Chem 1998;273:34406-12.
143. Chen CY, Melo E, Jakob P, et al. N-Terminomics identifies HtrA1 cleavage of thrombospondin-1 with generation of a proangiogenic fragment in the polarized retinal pigment epithelial cell model of age-related macular degeneration. Matrix Biol 2018;70:84-101.
144. Yang Z, Tong Z, Chen Y, et al. Genetic and functional dissection of HTRA1 and LOC387715 in age-related macular degeneration. PLoS Genet 2010;6:e1000836.
145. Zhang L, Lim SL, Du H, et al. High temperature requirement factor A1 (HTRA1) gene regulates angiogenesis through transforming growth factor-β family member growth differentiation factor 6. J Biol Chem 2012;287:1520-6.
146. Kanda A, Stambolian D, Chen W, Curcio CA, Abecasis GR, Swaroop A. Age-related macular degeneration-associated variants at chromosome 10q26 do not significantly alter ARMS2 and HTRA1 transcript levels in the human retina. Mol Vis 2010;16:1317-23.
147. Wang G, Scott WK, Haines JL, Pericak-Vance MA. Genotype at polymorphism rs11200638 and HTRA1 expression level. Arch Ophthalmol 2010;128:1491-3.
148. Wang G, Dubovy SR, Kovach JL, et al. Variants at chromosome 10q26 locus and the expression of HTRA1 in the retina. Exp Eye Res 2013;112:102-5.
149. Zurawa-Janicka D, Kobiela J, Stefaniak T, et al. Changes in expression of serine proteases HtrA1 and HtrA2 during estrogen-induced oxidative stress and nephrocarcinogenesis in male Syrian hamster. Acta Biochim Pol 2008;55:9-19.
150. Supanji Shimomachi M, Hasan MZ, Kawaichi M, Oka C. HtrA1 is induced by oxidative stress and enhances cell senescence through p38 MAPK pathway. Exp Eye Res 2013;112:79-92.
151. Lu Z, Lin V, May A, et al. HTRA1 synergizes with oxidized phospholipids in promoting inflammation and macrophage infiltration essential for ocular VEGF expression. PLoS One 2019;14:e0216808.
152. Tsuchiya A, Yano M, Tocharus J, et al. Expression of mouse HtrA1 serine protease in normal bone and cartilage and its upregulation in joint cartilage damaged by experimental arthritis. Bone 2005;37:323-36.
153. Hadfield KD, Rock CF, Inkson CA, et al. HtrA1 inhibits mineral deposition by osteoblasts: requirement for the protease and PDZ domains. J Biol Chem 2008;283:5928-38.
154. Lin MK, Yang J, Hsu CW, et al. HTRA1, an age-related macular degeneration protease, processes extracellular matrix proteins EFEMP1 and TSP1. Aging Cell 2018;17:e12710.
155. Grau S, Richards PJ, Kerr B, et al. The role of human HtrA1 in arthritic disease. J Biol Chem 2006;281:6124-9.
157. Schlecht A, Leimbeck SV, Jägle H, Feuchtinger A, Tamm ER, Braunger BM. Deletion of Endothelial Transforming Growth Factor-β Signaling Leads to Choroidal Neovascularization. Am J Pathol 2017;187:2570-89.
158. Fritsche LG, Igl W, Bailey JN, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet 2016;48:134-43.
159. Hollborn M, Stathopoulos C, Steffen A, Wiedemann P, Kohen L, Bringmann A. Positive feedback regulation between MMP-9 and VEGF in human RPE cells. Invest Ophthalmol Vis Sci 2007;48:4360-7.
160. Austin BA, Liu B, Li Z, Nussenblatt RB. Biologically active fibronectin fragments stimulate release of MCP-1 and catabolic cytokines from murine retinal pigment epithelium. Invest Ophthalmol Vis Sci 2009;50:2896-902.
161. Rosenfeld PJ, Brown DM, Heier JS, et al; MARINA Study Group. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 2006;355:1419-31.
162. Falavarjani KG, Nguyen QD. Adverse events and complications associated with intravitreal injection of anti-VEGF agents: a review of literature. Eye (Lond) 2013;27:787-94.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lu ZG, May A, Dinh B, Lin V, Su F, Tran C, Adivikolanu H, Ehlen R, Che B, Wang ZH, Shaw DH, Borooah S, Shaw PX. The interplay of oxidative stress and ARMS2-HTRA1 genetic risk in neovascular AMD. Vessel Plus 2021;5:4. http://dx.doi.org/10.20517/2574-1209.2020.48
AMA Style
Lu ZG, May A, Dinh B, Lin V, Su F, Tran C, Adivikolanu H, Ehlen R, Che B, Wang ZH, Shaw DH, Borooah S, Shaw PX. The interplay of oxidative stress and ARMS2-HTRA1 genetic risk in neovascular AMD. Vessel Plus. 2021; 5: 4. http://dx.doi.org/10.20517/2574-1209.2020.48
Chicago/Turabian Style
Lu, Zhi-Gang, Adam May, Brian Dinh, Victor Lin, Fei Su, Christina Tran, Harini Adivikolanu, Rachael Ehlen, Briana Che, Zhi-Hao Wang, Daniel H. Shaw, Shyamanga Borooah, Peter X. Shaw. 2021. "The interplay of oxidative stress and ARMS2-HTRA1 genetic risk in neovascular AMD" Vessel Plus. 5: 4. http://dx.doi.org/10.20517/2574-1209.2020.48
ACS Style
Lu, Z.G.; May A.; Dinh B.; Lin V.; Su F.; Tran C.; Adivikolanu H.; Ehlen R.; Che B.; Wang Z.H.; Shaw DH.; Borooah S.; Shaw PX. The interplay of oxidative stress and ARMS2-HTRA1 genetic risk in neovascular AMD. Vessel Plus. 2021, 5, 4. http://dx.doi.org/10.20517/2574-1209.2020.48
About This Article
Copyright

Data & Comments
Data


 Cite This Article 14 clicks
Cite This Article 14 clicks

 Like This Article 12
likes
Like This Article 12
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.