
Oxidative stress and inflammation in the development of cardiovascular disease and contrast induced nephropathy

Abstract
Utilization of contrast media to visualize vasculature structures in the setting of cardiovascular disorders (CVD) can lead to acute kidney injury, referred to as contrast-induced nephropathy (CIN). CIN can potentiate mortality and hospitalization in aged individuals, patients with CVD, nephropathy, enhancing kidney damage, and cardiac events. Preventing CIN by identifying risk factors is important. The underlying mechanisms of CIN pathology are unclear, but the key factors include direct cytotoxicity, oxidative stress, vascular and endothelial dysfunction and inflammatory processes. Reactive Oxygen Species and inflammatory mediators have been proposed as key factors influencing the development of CIN and CVD, and the elucidation of the interplay between the mechanisms evoked by them may provide a better understanding of the signaling processes happening in these conditions, thereby potentially enabling early identification, prevention and characterization of novel drug targets.
Keywords
Introduction
Inflammation is an immune system response to pathogenic insults and is physiologically important to protect the organism from injury. Inflammatory responses are triggered by harmful stimuli and lead to a removal of invading pathogens and initiation of the healing process[1]. Reactive oxygen species (ROS) modulate the inflammatory processes[2-5]. ROS include chemically heterogeneous free radicals (e.g., superoxide) and non-radicals (e.g., hydrogen peroxide) vital for cell development, survival and signaling[6]. Redox signaling occurs through posttranslational oxidation of proteins (e.g., cysteine residues)[7,8]. Moreover, there is also a known cross-talk between ROS and neutrophil inflammation clearance and pro-inflammatory markers[5,9]. Usually, these mechanisms are tightly regulated and when sustained and aberrant, inflammatory responses and ROS can lead to tissue damage and disease.
Environmental stress can cause oxidative stress, often defined by cell/tissue injury and attendant oxidative macromolecule damage[10]. Moreover, ROS have been highlighted as a cause of several inflammatory diseases like cardiovascular diseases (CVD), type II diabetes and cancer.
Due to its role promoting inflammation and lipid peroxidation, ROS have been tightly linked to CVD[11]. Thus, both inflammatory elements and ROS are CVD risk factors, described as underlying participants in the progression of atherogenesis. In addition, chronic inflammatory diseases, characterized by an involvement of oxidative stress in their pathogenesis, promote high risk and influence CVD susceptibility[12,13]. Inflammatory molecules and ROS have been proposed as possible predictors and drug targets in CVDs, reviewed by Cervantes Gracia, Llanas-Cornejo, & Husi, 2017[14,15]. Interestingly, target organ damage, described as the strong association with high blood pressure and functional changes in the heart, brain, eyes and kidney, is known to have significant implications in CVD onset[16,17]. Furthermore, CVD is a characteristic hallmark of severe kidney failure. Patients with chronic kidney disease (CKD) have been well characterized to carry a significantly higher risk of developing and dying from severe CVDs[18-20]. Therefore, management of chronic kidney disease progression has been proposed as strategy to reduce the incidence of cardiovascular events[21]. Conversely, the presence of CVDs have also been associated with a higher risk of renal impairment and CKD progression[22]. However, the influence that one disease has over the other, as well as the underlying molecular mechanisms remain to be elucidated.
To add to this pathology, kidney failure exacerbated by coronary intervention procedures relying on contrast media (CM), known as contrast induced nephropathy (CIN), constantly increases the incidence of comorbidities in this group of patients undergoing interventions and its prevention is challenging[23-26]. Since pre-existing CKD is the most common cause of CIN[27,28], the interplay among the underlying mechanisms of CVD and kidney failure are important. Additionally, inflammation and ROS have been identified as risk factors of CIN and as potential targets for prophylaxis or treatment[29-34]. Hence, the elucidation of CIN/CVD interplay in this setting would improve understanding of the signaling processes and progression of the diseases, leading the way to different approaches to either early detection or to identification of novel drug targets.
CIN pathophysiology in the context of CVD
According to the WHO, non-communicable diseases (NCD) account for 71% of all deaths world-wide and CVDs are responsible for most NCD deaths. CVDs were responsible for about 17.8 million deaths in 2017[35,36], and are the primary cause of death globally. Notably, angioplasty is the most common percutaneous coronary intervention (PCI) method for CVD treatment[37,38], and diagnostic angiography and PCI routinely utilize iodinated CM for vascular visualization[39,40]. Although angiograms and PCI can effectively diagnose and treat CVD patients, this can potentially lead to acute kidney diseases such as CIN induced by CM[41-46]. CM can be retained by the kidney where they have the potential to cause toxicity, resulting in acute renal injury[47]. Alternative CM have been developed to perform these procedures, but patients with risk factors such as kidney malfunction, diabetes, advanced age, CVD, anemia and hypotension are at high-risk and remain vulnerable to CIN[48,49].
CIN is a reversible form of acute renal injury that becomes evident after 48-72 h of intravascular administration of iodinated CM, manifesting in an increase of at least 25% in the serum creatinine level from baseline[50-52]. Although CIN can be transient, and in most of the cases serum creatinine level normalizes in 5-10 days, it can be irreversible and is associated with increased mortality and morbidity[25,45,50,53,54]. CIN is known to increase hospitalization, cardiovascular events, hepatic failure, dialysis and cardiac mortality, thus being directly associated with detrimental cardiac outcomes[50,55-57]. Additionally, CIN is attributable for a third of all hospital-acquired acute kidney injuries and its incidence can be as high as 50% in high-risk patients undergoing any procedure relying on intravascular contrast[26,47,58-60]. Regarding CIN treatment, it is limited and mainly supportive[60], thus early identification of at-risk patients is crucial and can be a potential approach for its management.
Although the precise pathophysiology of CIN is incompletely understood, crucial mechanisms have been associated with CIN, such as vasoconstriction in the renal vasculature, oxidative stress, renal medullary hypoxia, direct renal tubular cytotoxicity, and viscosity[45,53,61][Figure 1]. It has been proposed that the interplay of cytotoxicity and viscosity caused by CM, may be key in CIN pathophysiology. CM causes damage and apoptosis in surrounding endothelial cells (EC) and tubules of the nephron through iodine[62,63]. Moreover, vasoconstriction is known to increase blood viscosity after CM administration. CM increased viscosity and tubular pressure, exacerbating renal hypoperfusion and promote a decrease in urine flow rate, leading to its retention and allowing its continuous cytotoxicity [Figure 1][64,65]. Furthermore, blood viscosity is a key player in CVD pathophysiology and is associated with increased risks of CVD[66,67]. It has also been reported in the context of renal dysfunction and is associated with an increased risk of CVD and CKD development[68].
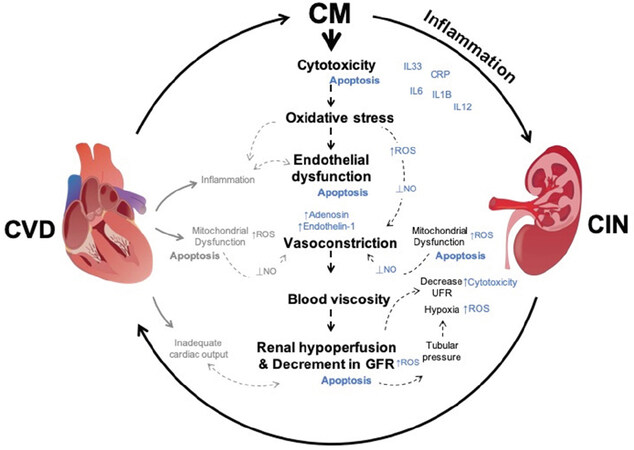
Figure 1. Contrast induced nephropathy and cardiovascular disorders pathophysiology interaction. Mechanisms triggered by Contrast Media (represented in black) lead to CIN. Their interaction promotes mitochondrial dysfunction, excessive ROS, promotes cytotoxicity and apoptosis. CIN enhances CVD pathophysiology. CVD processes (represented in grey) show CIN/CVD potential interaction creating a feedback loop that will enhance heart and kidney malfunction. Outcome from CM induced processes are represented in blue. ⟂: repression/reduction; ↑: overproduction; ROS: reactive oxygen species; NO: nitric oxide; UFR: urine flow rate; IL: interleukin; CRP: C-reactive protein; CIN: contrast induced nephropathy
Vasoconstrictor mediators (endothelin, adenosine, angiotensin II, vasopressin) are known to play a key role CIN and CVD pathogenesis[65,69-73]. CM is known to cause immediate vasoconstriction and vasodilation impairment, reduce renal blood flow, decrease glomerular filtration rate (GFR) and cause renal hypoperfusion, which leads to an inadequate delivery of oxygen, promoting ischemic injury [Figure 1]. These processes are associated with oxidative stress promoted by CM[74,75]. Decrease in GFR has also been associated with increased risk in CVD mortality, a feature that reflects kidney damage[76]. Regarding vasodilatation impairment, it has been suggested to be induced by CM through decreased nitric oxide (NO) bioavailability. This event has been proposed to be a result of loss of vasoactive NO and cellular damage on account of generation of peroxynitrite (ONOO-). Under physiological conditions, ROS production is attributed to nephron tubular transport regions with dense mitochondria populations, an important source of ROS[77,78]. Additionally, mitochondrial dysfunction is a key player in acute kidney injury[79,80] and is a characteristic feature of ageing, and chronic diseases, including diabetes and CVD, which are considered to be major risk factors for CIN[81,82]. Mitochondria are also abundant within cardiac cells due to the high energy demands, and notably mitochondrial ROS production is associated with CVD development[15,83]. Deleterious events such as arterial hypertension, endothelial dysfunction, atherosclerotic plaque formation and heart failure are associated with mitochondrial dysfunction[84-86]. Mitophagy removes damaged mitochondria and its impairment is a feature in CVD development as well. Moreover, ROS can induce damage in mitochondrial DNA, and damaged mitochondria are important sources of ROS; therefore, ROS overproduction due to mitophagy impairment disturbs homeostasis and leads to inflammation and apoptosis[87,88]. As in CIN, excessive ROS production from mitochondria is also associated with NO vasodilator impairment and it is tightly linked with endothelial dysfunction in cardiac event[89]. To add to CIN pathophysiology, oxygen imbalance under hypoxic conditions also leads to ROS production by the conversion of adenosine triphosphate (ATP) into hypoxanthine and its further reduction by xanthine oxidase. Mitochondrial dysfunction is also responsible for reduction in ATP synthesis, and will add to the cellular apoptotic state [Figure 1]. Interestingly, it has been recently reported that CKD in a rat model can influence cardiac pathologies by changing the function of cardiac tissue and inducing mitochondrial swelling and damage[90].
Inflammation is also a CIN hallmark, since the mechanisms previously described can trigger inflammatory processes. Notably, the presence of inflammatory elements has also been set as a feature for the population at high risk of CIN. Several studies have reported that the presence of active inflammatory processes biomarkers in patients with CVD may attribute its high-risk to developing CIN after CM exposure[30,31,91-94].
Cardiac insufficiency is also accountable for renal function impairment, emphasizing the complex interactions between heart and kidney where dysfunction in one organ can result in injury of the other[95]. Since CVD is a high-risk factor in CIN, and CIN can exacerbate CVD mortality, it is important to identify potential biomarkers for early detection and development of appropriate treatments. CIN processes that induce the release of vasoconstrictors, ROS and inflammatory cytokines have also been defined as hallmarks in CVDs due to the promotion of myocardial damage[50,96,97]. Additionally, a drastic decline in renal function may accelerate cardiovascular impairment by triggering inflammatory pathways[95,98]. Although an association of these events has been suggested for many years, its interplay remains to be described. Elucidating the possible interplay between oxidative stress and inflammation is important.
Oxidative stress in CVD and CIN
ROS play a significant role as second messengers within cells and regulate normal cellular functions, including gene transcription, signal transduction and homeostasis[99]. Many sources of ROS exist within cells and amongst ROS, the free radical superoxide (O
Mitochondria are responsible for the bulk ATP synthesis via chemiosmotic oxidative phosphorylation (OXPHOS). OXPHOS involves mobile electron carriers shuttles (NADH, cytochrome C and Coenzyme Q), protein complexes (complexes I-IV and the ATP-synthase complex) and a sequence of redox reactions where electrons are transported across the complexes of the respiratory chain up to complex IV, where molecular oxygen is reduced to water. The proton pumps establish an electrochemical proton motive force necessary for OXPHOS. Mitochondrial ROS can directly disturb the functionality of the ETC complexes by oxidizing iron-sulfur clusters and protein thiols [Figure 2][101-103]. Although mitochondria are a major source for ROS production, no clinical studies have been reported for mitochondrial-targeted antioxidants. This is largely due to the complications surrounding the targeted antioxidant delivery of injured mitochondria. Another cause for concern is that the role of mitochondrial ROS differs from cytosolic ROS as they are responsible for intracellular functions, which are maintained at a delicate equilibrium that could be negatively influenced by the careless use of antioxidants[104].
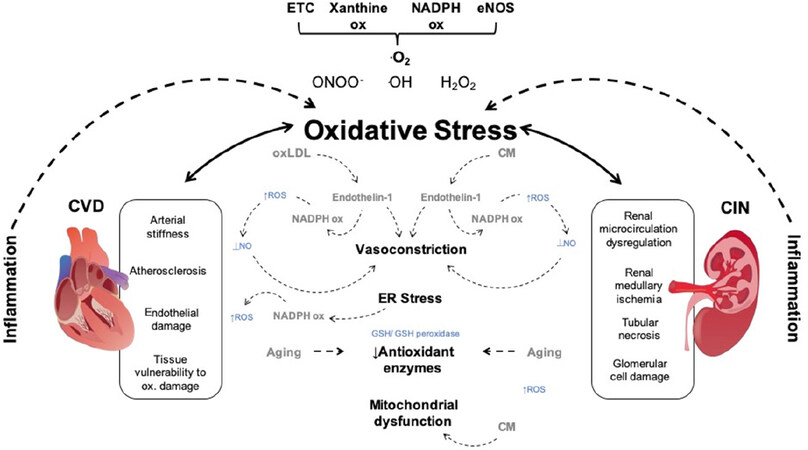
Figure 2. Oxidative stress mechanisms in contrast induced nephropathy and cardiovascular disorders. ONOO-, OH, O
It is understood that ageing is associated with cardiovascular oxidative stress[105]; tissue vulnerability to oxidative damage and is likely to be a key contributor in the development of cardiovascular disease[106]. Direct CM-induced toxicity on renal tubular epithelial cells appears to be a major contributing factor in CIN. CM induces renal vasoconstriction, through increased adenosine and endothelin-1 secretion, and diversion of blood flow from the medulla to the cortex [Figure 2]. Consequently, renal blood flow to the medulla and GFR is reduced, followed by ischemia in the renal medulla[107,108].
Atherosclerosis is the main cardiovascular disorder in which the association with oxidative stress became evident. Oxidized low density lipoprotein (oxLDL) plays a critical role in the pathogenesis of atherosclerosis. Studies have shown a clear link between arterial stiffness and oxLDL concentration, independent of the typical CVD risk factors[109]. It remains uncertain whether oxLDL as an oxidative stress biomarker has any predictive property in cardiovascular patients[110].
Vascular NOXs are important ROS generating enzymes and in human vascular cells, NOX1, NOX2, NOX4 and NOX5 are expressed. NOX are transmembrane enzyme complexes with a few regulatory subunits and a core catalytic subunit, except for NOX5[111]. NOX activation results in the generation of O
NOX4 plays a key regulatory role, generating athero-protective ROS that inhibits inflammation and vascular remodeling. Decreased levels of effector T cells and chemokines, increased regulatory T-cells and reduced lesion formation was seen in apolipoprotein E-deficient mice expressing ectopic endothelial NOX4[114]. However, reduced levels of endothelial H
As well as the increased production of oxLDL, an additional contributor to cardiovascular morbidity appears to be oxidative endothelial damage. In healthy adults of varying ages, brachial artery flow-mediated dilation appeared to inversely correlate with the concentration of nitrotyrosine (produced, for example, via nitrogen dioxide radical and tyrosine radical recombination) in vascular EC[119]. ET-1, as well as being a powerful vasoconstrictor, has also demonstrated proinflammatory and prooxidant properties and consequently, it has been associated with oxidative endothelial damage[120]. In EC, oxLDL has been shown to stimulate endothelin-1 production, and elevated levels of endothelin-1 is known to generate ROS by NADPH oxidase [Figure 2][121]. Furthermore, the cardiovascular system inflammatory response is induced by oxidative stress and proinflammatory cytokines additionally induce oxidative damage in a positive, reverse feedback mechanism [Figure 2][122].
Antioxidant defense mechanisms decrease with age[123], therefore age is a major risk factor of CIN. The unique anatomy of the renal medulla requires the thick ascending limbs of the loop of Henle to carry out energetically challenging ion transport in a state of relative hypoxia compared to the renal cortex. It has been proposed that a discrepancy between the metabolic requirements of these thick ascending limbs and the medullary blood supply could generate O
A crucial factor in the production of ROS in the kidney is renal hypoxia. There are, however, conflicting reports relating to the extent to which oxidative stress is a cause or epiphenomena. ROS are regularly involved in cellular inflammatory responses and it is proposed that ROS are formed during renal parenchymal hypoxia, following CM exposure, resulting in vascular endothelial injury. This aggravates renal parenchymal hypoxia resulting in endothelial dysfunction[125]. O
In response to excessive oxidative stress, cells activate/induce their own antioxidant defense mechanisms. Glutathione (GSH), is an important endogenous thiol that is essential to a variety of detoxification processes. Mammalian cells contain high concentrations of GSH (3-5 mmol/L) which is used in numerous diverse roles as well as hepatic detoxification. GSH can donate reducing equivalents for the activity of specific antioxidant peroxidase enzyme, such as GSH peroxidase (GPx), and can react directly with certain ROS (e.g., carbonate radical). Intracellular levels of GSH are tightly controlled by the enzymes glutamate-cysteine ligase and GSH synthase (involved in synthesis), GSH reductase (involved in recycling of oxidized glutathione back to GSH) and GSH transferases (involved in utilization)[130]. Redox enzymes include thioredoxin, catalase, GPx, peroxiredoxins and superoxide dismutase (SOD)[100].
Role of inflammation in CIN/CVD
One of the factors that is central to the prevalence of CIN is chronic inflammation. The role of inflammation in CIN has been extensively studied and clinical trials in humans and animal models have been performed to help elucidate this role[131-134]. One of the main features of intravascular iodinated CM is that it causes vasodilation followed by a prolongation in vasoconstriction[135,136]. The vasodilation/vasoconstriction occurs in all patients that require a CM procedure, but this effect has not been found to work alone in the increase of CIN risk among patients. Two additional pathways suggested to promote this increase are cellular toxicity and elevated urinary viscosity that can cause obstructions through stone formation[137].
Although the global prevalence of CIN does not constitute a public health threat, at risk populations, such as those suffering from higher presence of infectious diseases, have a higher incidence of inflammation than populations that are not affected by these diseases[138]. A close relationship between inflammatory molecules and thrombogenesis has been well reported[139]. The acute inflammatory state is a landmark of infectious diseases and one of the main type of molecules that derive from it are interleukins (ILs). IL-6 and IL-12 have been targeted as disruptors of homeostasis within inflammatory processes. IL-6 promotes the expression of the C reactive protein (CRP), which is being used as a current acute inflammation marker [Figure 3][140].
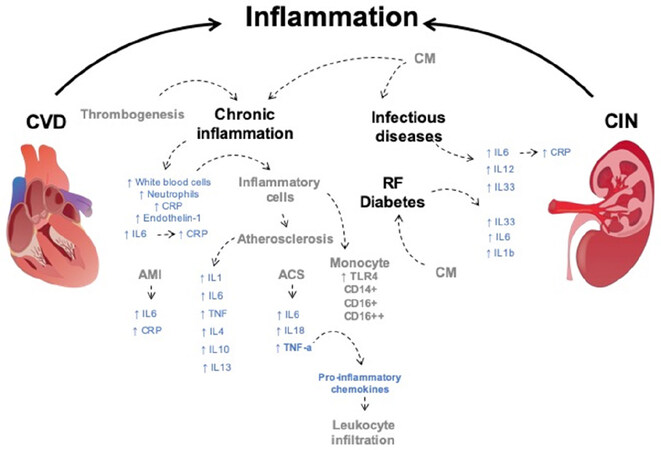
Figure 3. Inflammatory molecules in CIN and CVD. Inflammatory states have been associated with CIN and CVD risk factors. Inflammatory cells and molecules are considered as potential risk factors in CVD and CIN. Inflammatory risk factors highlighted in blue. CM, Disease states and cellular types related to inflammatory risk factors represented in grey. ↑: overproduction. CM: contrast media; RF: risk factors; AMI: acute myocardial infarction; ACS: acute coronary syndrome; IL: interleukin; CRP: C reactive protein; TNF-α: tumor necrotic factor-α; TLR4: toll like receptor 4; CIN: contrast induced nephropathy; CVD: cardiovascular disorders
One of the studies that assessed the increased risk for CIN due to inflammation was performed by Kwasa et al.[132]. They performed a prospective cohort study of patients undergoing a contrast-enhanced CT (CECT) scan. 423 patients were recruited and grouped into those without inflammation having serum CRP levels ≤ 5mg/dL and those with evidence of inflammation having CRP levels > 5 mg/dL. Serum creatinine (SCr) was measured before the CECT and 48 h following the CECT with CIN diagnosed by an increase of > 25% in SCr from the baseline [Figure 3]. The observed incidence of CIN was 9.92%. Of the patients with inflammation, 29 (13.5%) developed CIN, while 13 (6.25%) of those without inflammation developed CIN. No significant relation was found between the increase of CIN prevalence and biophysical variables (age, sex, height, weight, etc.)[132]. Another study reported by Oweis et al.[30] showed serum levels of IL-33 as significant predictor for development of CIN. Of the total 202 patients, 30 (14.8%) developed CIN. The incidence rate was 21.1% among females and 12.4% among males [Figure 3].
Additional biomarkers of inflammation have been studied to assess their potential as predictors of CIN in different conditions. Cell types that are associated chronic inflammation have been proposed as predictors of increased risk of developing CIN: the study published by Yuan et al.[92] in 2017 found in 1,061 patients that white blood cell count, neutrophil count, neutrophil lymphocyte ratio, CRP level, and big ET-1 level were all associated with an increased risk of CIN development. It is important to mention that all of the patients in this study went through emergency PCI.
Regarding the assessment of multiple markers to predict the development of CIN, different studies have reported combinations between proteins that can be measured in human serum. The study performed by Satilmis et al.[141], presented an assessment of the ratio between 2 inflammatory markers, CRP and albumin. 205 patients with non-ST-elevation myocardial infarction that underwent PCI were subsequently assessed for development of CIN. The prevalence of CIN in this study was 10.2%. Multivariate logistic regression analysis showed significant association between CRP: albumin ratio and the development of CIN; advanced age, diabetes, dyslipidemia and left ventricular ejection fraction were also associated with the condition.
Animal models have also been used in the search for the potential role of inflammation in the development of CIN. Demirtas et al.[29] evaluated the role of IL-33 in the pathogenesis of CIN in diabetic rats. 30 male Sprague-Dawley rats were divided into 3 groups (healthy, diabetic and diabetic with CIN). Significantly increased presence of IL-33 was found in the kidney tissue of the diabetic group after induction of CIN when compared with the healthy and diabetic groups. Serum levels of IL-33, IL-6, and IL-1β were also significantly increased in the diabetic + CIN group when compared to the healthy and diabetic groups [Figure 3].
Prophylactic use of carotenoids has been studied in animal models to assess the relation between oxidative stress induced inflammation and CIN development. The studies presented by Buyuklu et al.[142,143] aimed to investigate the effects of lycopene and curcumin as protection against the development of CIN in rats. 28 male Wistar albino rats were divided into 4 groups, they included a normal control group, CIN group, CIN + lycopene and CIN + curcumin groups. Significant increase in urea, creatinine and malondialdehyde were observed in the CIN group when compared with the control group. Additionally, histological tests showed significant increase of infiltrated inflammatory cells and necrotic degenerative changes in the CIN group when compared against the control[142,143].
The role of the inflammatory state in CVD was addressed in an extensive literature[14]. The search for markers has two principal aims: to look into the understanding of the mechanisms of disease and to identify molecules that can be detected more accurately to predict the risk of cardiovascular events. The role of inflammation in CVD development has been assessed throughout different populations and experimental models, critical importance has been given to events such as acute myocardial infarction (AMI) and atherosclerosis due to their high incidence and mortality rates[144]. Inflammation in CVD includes a vast number of processes which can occur at the site of disease, in the bloodstream and at sites far from the disease[145]. Immune response takes the spotlight when addressing inflammation and CVD. In AMI a signaling cascade induces the expression and recruitment of proinflammatory molecules, accelerating both damage and further repair of injured cardiac tissue. Elevated levels of high-sensitivity CRP and IL-6 in plasma have been found correlated with unfavorable outcomes in patients [Figure 3][146].
Rajendran et al.[147] assessed both IL-6 and hs-CRP in a Chennai based population. 93 patients with AMI and 102 healthy subjects as a control group were analyzed. Both IL-6 and hs-CRP were found to be significantly increased when compared with the control group. Pro-inflammatory cytokines IL-6, IL-10, IL-18 and TNF-α were evaluated in a study published in 2019 including 120 patients with acute coronary syndrome (ACS) and 60 healthy controls. Serum levels of IL-6, IL-18 and TNF-α were significantly higher in the ACS group when compared to the healthy group [Figure 3]. No significant difference in serum levels of IL-10 was found[148]. Additionally, TNF-α has been found to promote the release proinflammatory chemokines and adhesion molecule synthesis in damaged myocardium and causing additional leukocyte infiltration in mice[149].
Toll-like receptors (TLRs) may be key to understanding heart failure. TLR4 deficiency is associated with decreased in size of damage by infarct and reduction of systemic inflammation in mice[150]. In humans, the activation of TLR4 in monocytes is associated with the development of cardiac failure after AMI [Figure 3][151]. By contrast, deficiencies in the function of TLR2 were found to reduce myocardial fibrosis and improve ventricular remodeling after AMI in a murine model[152].
Atherosclerosis is often described as a chronic inflammatory process. Deregulation in the endothelium is mediated by cell adhesion molecules, such as ICAM1, P-selectin and VCAM1. Additionally, the secretion of cytokines has a role in atherogenesis, namely IL-1, IL-6, TNF, IL-4, IL-10 and IL-13 [Figure 3]. The detection of some of these molecules in plasma has identified associations that could help to predict atherosclerosis severity. Moreover, the identification of cell types through flow cytometry has proven to be a promising predictor for atherogenic levels of severity. The amount CD14+CD16++ monocytes present in circulation has been found to be inversely correlated to plasma HDL levels while CD16+ monocytes levels are proportional to severe atherosclerosis [Figure 3][153].
CVD and CIN biomarkers
The identification of rapid, predictive biomarkers for CIN is essential as current targets are relatively slow to be useful, or the assays are just too expensive to be launched in a clinical setting. Some of the postulated biomarkers for CIN and CVD are shown on Table 1. An early predictive biomarker of AKI is human neutrophil gelatinease-associated lipocalin (NGAL). NGAL is a small protein of the lipocalin superfamily that was initially identified from the supernatant of activated human neutrophils in 1993. Successive studies have recognized renal NGAL as a unique, specific biomarker for the early detection of AKI in critically ill patients and after CM administration. Urinary and serum levels of NGAL increase well before the increase of serum creatinine levels (~2 h). As a result, NGAL is increasingly studied as a marker of AKI[154-157]. Another proposed sensitive, early, non-invasive biomarker for AKI kidney injury is urinary neutrophil gelatinase-associated lipocalin (uNGAL) also known as lipocalin-2. uNGAL is an iron-transporting protein that rapidly accumulates in the urine and kidney tubules after nephrotoxic and ischemic insults. Zappitelli et al.[158] concluded that uNGAL is an effective predictor of AKI which is triggered in advance of increases in serum creatinine concentration. Despite these findings, the use of uNGAL is still experimental.
Origin and mechanisms of potential biomarkers for prediction of CIN and CVD
| Biomarkers | Etiology | Mechanisms | Organism | Ref. |
|---|---|---|---|---|
| IL-6, IL-12, IL-8 | CIN and CVD | Induction of the production of CRP | Human | Alladina et al.[171] (2016)
Kwasa et al.[132] (2014) Rajendran et al.[147] (2012) |
| C reactive protein | CIN and CVD | Response to chronic inflammation | Human | Kwasa et al.[132] (2014)
Rajendran et al.[147] (2012) |
| TNF-α | CVD | Upregulated in inflammation in acute myocardial infarction, modulates cardiac contractility and peripheral resistance. Promotes leukocyte infiltration in mice | Human
Mice | Senguttuvan et al.[148] (2019)
Maekawa et al.[149] (2002) |
| CD14+CD16++ monocytes | CVD | Presence inversely correlated to plasma HDL levels | Human | Schlitt et al.[153] (2004) |
| CD16+ monocytes | CVD | Levels proportional to severe atherosclerosis | Human | Schlitt et al.[153] (2004) |
| Neutrophil/Lymphocyte ratio | CIN | Elevated in subclinical inflammation | Human | Yuan et al.[92] (2017) |
| CRP/Albumin ratio | CIN | CRP levels are found increased in chronic inflammation and albumin levels are negatively correlated in the presence of acute inflammation | Human | Satilmis et al.[141] (2020) |
| IL-33 and IL-1β | CIN and CVD | Proinflammatory cytokines, IL-33 binds to immune cells and promotes secretion of cytokines resulting in inflammation | Human and Sprague-Dawley rat | Oweis et al.[30] (2018)
Demirtas et al.[29] (2016) |
| NGAL | CIN | Accumulates in urine, blood and renal cortical tubules following ischaemic and nephrotoxic injury. Antioxidant protection against CIN development | Human
Wistar albino rat | Malyszko et al.[156] (2009)
Buyuklu et al.[143] (2014) |
| L-FABP | CIN | Specifically binds to intracellular, free unsaturated fatty acids during hypoxic tissue injury | Human | Nakamura et al.[159] (2006) |
| tPA | CIN and CVD | Tissue type fibrinolytic agent involved in the breakdown of blood clots and the recruitment of inflammatory cells | Human | Baramova et al.[160] (1997) and Stringer et al.[161] (1997) |
| uPA | CIN and CVD | Urokinase type fibrinolytic agent involved in the breakdown of blood clots and the recruitment of inflammatory cells | Human | Baramova et al.[160] (1997) and Stringer et al.[161] (1997) |
| PAI-1 | CIN and CVD | Primary physiological inhibitor of tPA and uPA | Human | Baramova et al.[160] (1997) and Stringer et al.[161] (1997) |
| KIM-1 | CIN | Localised to the proximal tubules of the human kidney following toxic or ischaemic injury | Human | Nogare et al.[172] (2012) |
| IL-18 | CIN and CVD | Proinflammatory cytokine | Human
Mice | Ling et al.[168] (2008) |
| CysC | CIN | Produced by all nucleated cells and displays a stable rate of production. Freely filtered by the glomerulus | Human | Soto et al.[174] (2010) |
| Serum Creatinine | CIN | Resulting product of creatine phosphate from protein and muscle metabolism. Exhibits a stable rate of production and is freely filtered by the glomerulus | Human | Slocum et al.[173] (2012) |
Liver type fatty acid binding protein (L-FABP) is an intracellular lipid chaperone and is expressed in renal proximal tubule cells and secreted into the urine in response to hypoxia caused by a decrease in peritubular capillary blood flow. Although L-FABP concentration is significantly increased in CIN patients after 24 hours, the specificity of this biomarker for CIN is low on account of a range of potential confounders[159].
Tissue plasminogen activator (tPA), a part of the serine protease family, is a plasma protein involved in the breakdown of blood clots and a key fibrinolytic agent that takes part in the recruitment of inflammatory cells. Some other roles of tPA involve the turnover of extracellular matrix components via activation of matrix metalloproteinases and immune-modulatory functions. Plasminogen activator inhibitor-1 is the main physiological inhibitor of endogenous fibrinolysis which functions through the inhibition of tPA and the urokinase type activator (uPA)[160,161]. A recent study[162] reported a relationship between increased serum tPA levels with an increased rate of mortality of dialysis-dependent AKI (AKI-D) patients. Elevated tPA expression has been detected in the proximal tubular epithelial cells of ischemic kidneys, in animal models. Removing tPA by antisense treatment had reduced the influx of neutrophils and helped protect renal function during ischemia-reperfusion injury. This suggests tPA inhibition as a novel strategy to improve ischemic AKI[163]. Many additional studies have also implied the involvement of tPA in the process of kidney fibrosis that leads to progression of CKD[164-166].
IL-6 is an interleukin that can act as both an anti-inflammatory myokine and a pro-inflammatory cytokine and is encoded by the IL6 gene in humans. Osteoblasts produce and release IL-6. The role of IL-6 role as an anti-inflammatory cytokine is facilitated via the interleukins inhibitory effects on IL-1 and TNF-α, and activation of IL-10 and IL-1ra[167]. Studies have demonstrated a close correlation between AKI and IL-6 expression in many animal models[168,169]. Resident kidney cells, such as tubular epithelial cells, endothelial cells, mesangial cells and podocytes can all produce and release IL-6. A study has shown that, in a model of ischemia-reperfusion injury, after leukocytes penetrated the injured kidney, maladaptive IL-6 was produced in response to their TLR-4 receptors interacting with high mobility group box 1 protein released by the injured renal cells[170]. Raised levels of the pro-inflammatory cytokines, IL-8 and IL-6, have been seen early on in AKI patients and were linked to prolonged mechanical ventilation[171].
The transmembrane protein, kidney injury molecule 1 (KIM-1), recognizes apoptotic cells and leads them to lysosomes. Additionally, it acts as a receptor for oxidized lipoproteins and is therefore adept at recognizing apoptotic cell signals. KIM-1 is undetectable in normal kidney tissue but is highly expressed following toxic or ischaemic injury in differentiated proximal tubule epithelial cells from rodent and human kidneys[172,173]. Plasma cystatine-C (CysC), is a low molecular weight protein produced at a predictable rate by all nucleated cells. CysC is filtered across the glomerular membrane but is neither reabsorbed nor secreted during its passage through the nephron. Given that CysC is almost entirely catabolized in the proximal tubule, it is impossible to measure its renal clearance. However, the plasma or serum concentration of CysC accurately reflects the GFR and significant increases in CysC are detected in CIN patients after 8 h. However, a similar increment has also been seen in several other conditions, including thyroid dysfunction, age, an increase in muscle mass, systemic inflammation, corticosteroids administration and neoplasia[174] limiting its utility as a CIN biomarker.
The key diagnostic criterion for CIN is the elevation of serum creatinine concentration by more than 25% over baseline, after eliminating any other possible causes. Other laboratory findings may also be present such as hyperkalaemia and acidosis. Although patients may have normal urine output, they can also suffer from anuria (failure of the kidneys to produce urine) or oliguria (low output of urine > 80 mL/day, < 400 mL/day). Findings on urine analysis are normally non-specific[175]. Normally a delay of 24-48 h is seen between contrast exposure and changes in serum creatinine concentration, which makes creatinine a late indicator of renal function changes[176].
Since a close correlation among inflammatory molecules and kidney injury in CIN has been observed, as described above, they have also been proposed as potential CIN biomarkers [Table 1]. IL-8 and IL-6, have been seen early on in AKI patients and were linked to prolonged mechanical ventilation[171]. Successive studies have recognized renal NGAL as a unique, specific biomarker for the early detection of AKI in critically ill patients and after CM administration[154-157]. Other proposed biomarkers, despite being effective predictors of AKI, such as uNGAL triggered preceding increases in serum creatinine concentration[157,158] are still experimental. Other potential biomarkers have been deemed as non-specific, such as L-FABP, although significantly increased in CIN patients after 24 h, where potential confounders lower its specificity[159].
Conclusion
Oxidative stress influences cardiovascular morbidity mainly through increased peripheral vascular resistance [Figure 1]. However, although the generation of ROS could affect renal blood flow by facilitating the production of vasoconstrictors and impacting the effects of vasodilators, the influence of oxidative stress in the development of CIN is uncertain.
Inflammation results in the alteration of homeostasis in both the circulatory and renal systems. These alterations can be intrinsic of cellular damage or can be mediated by external factors such as CM. Immune response to CM cytotoxicity causes a rapid increase in the migration and accumulation of cytokines such as ILs and TNF-α in the progression of both CVD and CIN. Additionally, the presence of cellular types found in response to inflammation is a feature in early development of CVD and CIN. The main interplay between CIN and CVD in the context of inflammation may rely on endothelial dysfunction and immune response. The signaling pathways activated through endothelial dysfunction in cardiac events result in the generation of systemic inflammation which has been found to affect the kidneys and made them more susceptible to local inflammation processes driven by CM cytotoxicity.
Current CIN prevention strategies, such as the use of carotenoids, for instance curcumin and lycopene[142,143], to limit the oxidative effects of CM are questionable due to the inconclusive evidence to support the oxidative capacity of CM. Existing biomarkers for CIN are either non-specific, such as L-FABP, or late indicators of renal function changes, such as changes in serum creatinine, making them poor predictive markers at best. The relationship between CVD and CIN and the underlying mechanisms responsible for CIN are unclear. Identifying novel biomarkers, be it genetic, redox or serum protein markers, for the early detection of CIN will help gain a better understanding of the underlying mechanisms. Greater mechanistic understanding is required to better predict and treat CIN.
Declarations
Authors’ contributionsDesign and conception of the original draft: Cervantes-Gracia K, Raja K, Llanas-Cornejo D
Original draft text editing: Cervantes-Gracia K, Raja K, Llanas-Cornejo D, Cobley JN, Megson IL, Chahwan R, Husi H
Figures design and editing: Cervantes-Gracia K
Table design and editing: Raja K, Llanas-Cornejo D
Reference list editing: Llanas-Cornejo D, Husi H
Led the study: Husi H
Availability of data and materialsNot applicable.
Financial support and sponsorshipCervantes-Gracia K is supported by CONACYT Mexico scholarship (No. 2019-000021-01EXTF-00542); Raja K is supported by the European Union’s INTERREG VA Programme, managed by the Special EU Programmes Body (SEUPB); Cobley JN and Husi H are supported by a grant from Highlands & Islands Enterprise.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Chen L, Deng H, Cui H, Fang J, Zuo Z, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018;9:7204-18.
2. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxidants Redox Signal 2014;20:1126-67.
3. Kim YM, Kim SJ, Tatsunami R, Yamamura H, Fukai T, et al. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am J Physiol - Cell Physiol 2017;312:C749-64.
4. Chelombitko MA. Role of reactive oxygen species in inflammation: a minireview. Moscow Univ Biol Sci Bull 2018;73:199-202.
5. Yang W, Tao Y, Wu Y, Zhao X, Ye W, et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat Commun 2019;10:1076.
7. Cai Z, Yan LJ. Protein oxidative modifications: beneficial roles in disease and health. J Biochem Pharmacol Res 2013;1:15-26.
8. Cobley JN, Husi H. Immunological techniques to assess protein thiol redox state: opportunities, challenges and solutions. Antioxidants 2020;9:31.
9. Ranneh Y, Ali F, Akim AM, Hamid HA, Khazaai H, et al. Crosstalk between reactive oxygen species and pro-inflammatory markers in developing various chronic diseases: a review. Appl Biol Chem 2017;60:327-38.
10. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 2010;49:1603-16.
11. Austin V, Crack PJ, Bozinovski S, Miller AA, Vlahos R. COPD and stroke: are systemic inflammation and oxidative stress the missing links? Clin Sci 2016;130:1039-50.
12. Rovira-Llopis S, Rocha M, Falcon R, de Pablo C, Alvarez A, et al. Is myeloperoxidase a key component in the ROS-induced vascular damage related to Nephropathy in type 2 diabetes? Antioxidants Redox Signal 2013;19:1452-8.
13. Hansen PR. Chronic inflammatory diseases and atherosclerotic cardiovascular disease: innocent bystanders or partners in crime? Curr Pharm Des 2018;24:281-90.
14. Lorenzatti AJ, Servato ML. New evidence on the role of inflammation in CVD risk. Curr Opin Cardiol 2019;34:418-23.
16. Williams B, Mancia G, Spiering W, Rosei EA, Azizi M, et al. 2018 ESC/ESH Guidelines for themanagement of arterial hypertension. Eur Heart J 2018;39:3021-104.
17. Vasan RS, Short MI, Niiranen TJ, Xanthakis V, DeCarli C, et al. Interrelations between arterial stiffness, target organ damage, and cardiovascular disease outcomes. J Am Heart Assoc 2019;8:e012141.
18. Briasoulis A, Bakri GL. Chronic kidney disease as a coronary artery disease risk equivalent. Curr Cardiol Rep 2013;15:340.
19. Yuan J, Zou XR, Han SP, Cheng H, Wang L, et al. Prevalence and risk factors for cardiovascular disease among chronic kidney disease patients: results from the Chinese cohort study of chronic kidney disease (C-STRIDE). BMC Nephrol 2017;18:23.
20. Tomey MI, Winston JA. Cardiovascular pathophysiology in chronic kidney disease: opportunities to transition from disease to health. Ann Glob Health 2014;80:69-76.
21. Subbiah AK, Chhabra YK, Mahajan S. Cardiovascular disease in patients with chronic kidney disease: a neglected subgroup. Heart Asia 2016;8:56-61.
22. Rahman M, Xie D, Feldman HI, Go AS, He J, et al. Association between chronic kidney disease progression and cardiovascular disease: results from the CRIC study. Am J Nephrol 2014;40:399-407.
24. Pyxaras SA, Sinagra G, Mangiacapra F, Perkan A, Di Serafino L, et al. Contrast-induced nephropathy in patients undergoing primary percutaneous coronary intervention without acute left ventricular ejection fraction impairment. Am J Cardiol 2013;111:684-8.
25. Sato A, Aonuma K, Watanabe M, Hirayama A, Tamaki N, et al. Association of contrast-induced nephropathy with risk of adverse clinical outcomes in patients with cardiac catheterization: from the CINC-J study. Int J Cardiol 2017;227:424-9.
26. Rear R, Bell RM, Hausenloy DJ, Hausenloy DJ. Contrast-induced nephropathy following angiography and cardiac interventions. Heart 2016;102:638-48.
27. Hossain MA, Costanzo E, Cosentino J, Patel C, Qaisar H, et al. Contrast-induced nephropathy: pathophysiology, risk factors, and prevention. Saudi J Kidney Dis Transpl 2018;29:1-9.
28. McDonald JS, McDonald RJ, Tran CL, Kolbe AB, Williamson EE, et al. Postcontrast acute kidney injury in pediatric patients: a cohort study. Am J Kidney Dis 2018;72:811-8.
29. Demirtas L, Turkmen K, Kandemir FM, Ozkaraca M, Kucukler S, et al. The possible role of interleukin-33 as a new player in the pathogenesis of contrast-induced nephropathy in diabetic rats. Ren Fail 2016;38:952-60.
30. Oweis AO, Alshelleh SA, Daoud AK, Smadi MM, Alzoubi KH. Inflammatory milieu in contrast-induced nephropathy: a prospective single-center study. Int J Nephrol Renovasc Dis 2018;11:211-5.
31. Yildirim E, Ermis E, Cengiz M. Inflammatory markers of contrast-induced nephropathy in patients with acute coronary syndrome. Coron Artery Dis 2020;31:279-83.
32. Murashima M, Nishimoto M, Kokubu M, Hamano T, Matsui M, et al. Inflammation as a predictor of acute kidney injury and mediator of higher mortality after acute kidney injury in non-cardiac surgery. Sci Rep 2019;9:20260.
33. de Souza Santos V, Peters B, Côco LZ, Alves GM, de Assis ALEM, et al. Silymarin protects against radiocontrast-induced nephropathy in mice. Life Sci 2019;228:305-15.
34. Kim JE, Bae SY, Ahn SY, Kwon YJ, Ko GJ. The role of nuclear factor erythroid-2-related factor 2 expression in radiocontrast-induced nephropathy. Sci Rep 2019;9:2608.
35. Kaptoge S, Pennells L, De Bacquer D, Cooney MT, Kavousi M, et al. World Health Organization cardiovascular disease risk charts: revised models to estimate risk in 21 global regions. Lancet Glob Health 2019;7:e1332-45.
36. Roth GA, Abate D, Abate KH, Abay SM, Abbafati C, et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018;392:1736-88.
37. Astin F, Jones K, Thompson DR. Prevalence and patterns of anxiety and depression in patients undergoing elective percutaneous transluminal coronary angioplasty. Heart Lung 2005;34:393-401.
38. Chhabra L, Zain MA, Siddiqui WJ. Angioplasty. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2020.
39. Nadolski GJ, Stavropoulos SW. Contrast alternatives for iodinated contrast allergy and renal dysfunction: options and limitations. J Vasc Surg 2013;57:593-8.
40. Schraeder R. Contrast media selection in interventional cardiology. J Clin Basic Cardiol 2001;4:245-8.
41. Al Shammeri O, Garcia LA. Thrombolysis in the age of primary percutaneous coronary intervention: mini-review and meta-analysis of early PCI. Int J Health Sci (Qassim) 2013;7:91-100.
42. Barauskas M, Unikas R, Tamulenaite E, Unikaite R. The impact of clinical and angiographic factors on percutaneous coronary angioplasty outcomes in patients with acute ST-elevation myocardial infarction. Arch Med Sci Atheroscler Dis 2016;1:e150-7.
43. Darvishpour A, Javadi-Pashaki N, Salari A, Sadeghi T, Taleshan-Nejad M. Factors associated with quality of life in patients undergoing coronary angioplasty. Int J Health Sci (Qassim) 2017;11:35-41.
44. Kim MJ, Jeon DS, Gwon HC, Kim SJ, Chang K, et al. Health-related quality-of-life after percutaneous coronary intervention in patients with UA/NSTEMI and STEMI: the Korean multicenter registry. J Korean Med Sci 2013;28:848-54.
45. Mandal A, Paudel MS, Kafle P, Khalid M, Bhattarai B, et al. Contrast-induced nephropathy following percutaneous coronary intervention at a tertiary cardiac center in Nepal. Cureus 2018;10:e3331.
46. Windecker S, Kolh P, Alfonso F, Collet JP, Cremer J, et al. 2014 ESC/EACTS Guidelines on Myocardial Revascularization: The Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS) Developed With the Special Contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J 2014;35:2541-619.
47. Manske CL, Sprafka JM, Strony JT, Wang Y. Contrast nephropathy in azotemic diabetic patients undergoing coronary angiography. Am J Med 1990;89:615-20.
48. Diamantopoulos A, Patrone L, Santonocito S, Theodoulou I, Ilyas S, et al. Carbon dioxide angiography during peripheral angioplasty procedures significantly reduces the risk of contrast-induced nephropathy in patients with chronic kidney disease. CVIR Endovasc 2020;3:9.
49. Ghumman SS, Weinerman J, Khan A, Cheema M, Levin D, et al. Contrast-induced nephropathy following peripheral angiography with carbon dioxide versus iodinated contrast media: a systematic review and meta-analysis of current literature. J Am Coll Cardiol 2017;69:2088.
50. Andreis A, Budano C, Levis M, Garrone P, Usmiani T, et al. Contrast-induced kidney injury: how does it affect long-term cardiac mortality? J Cardiovasc Med 2017;18:908-15.
51. Neyra JA, Shah S, Mooney R, Jacobsen G, Yee J, et al. Contrast-induced acute kidney injury following coronary angiography: a cohort study of hospitalized patients with or without chronic kidney disease. Nephrol Dial Transplant 2013;28:1463-71.
52. Schilp J, De Blok C, Langelaan M, Spreeuwenberg P, Wagner C. Guideline adherence for identification and hydration of high-risk hospital patients for contrast-induced nephropathy. BMC Nephrol 2014;15:2.
53. Haq MFU, Yip CS, Arora P. The conundrum of contrast-induced acute kidney injury. J Thorac Dis 2020;12:1721-7.
54. Marenzi G, Lauri G, Assanelli E, Campodonico J, De Metrio M, et al. Contrast-induced nephropathy in patients undergoing primary angioplasty for acute myocardial infarction. J Am Coll Cardiol 2004;44:1780-5.
55. Mehran R, Aymong ED, Nikolsky E, Lasic Z, Iakovou I, et al. A simple risk score for prediction of contrast-induced nephropathy after percutaneous coronary intervention: Development and initial validation. J Am Coll Cardiol 2004;44:1393-9.
56. Tsai TT, Patel UD, Chang TI, Kennedy KF, Masoudi FA, et al. Contemporary incidence, predictors, and outcomes of acute kidney injury in patients undergoing percutaneous coronary interventions: insights from the NCDR cath-PCI registry. JACC Cardiovasc Interv 2014;7:1-9.
57. Watabe H, Sato A, Hoshi T, Takeyasu N, Abe D, et al. Association of contrast-induced acute kidney injury with long-term cardiovascular events in acute coronary syndrome patients with chronic kidney disease undergoing emergent percutaneous coronary intervention. Int J Cardiol 2014;174:57-63.
58. Berwanger O. Acetylcysteine for prevention of renal outcomes in patients undergoing coronary and peripheral vascular angiography: Main results from the randomized acetylcysteine for contrast-induced nephropathy trial (ACT). Circulation 2011;124:1250-9.
59. Bolognese L, Falsini G, Schwenke C, Grotti S, Limbruno U, et al. Impact of iso-osmolar versus low-osmolar contrast agents on contrast-induced nephropathy and tissue reperfusion in unselected patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Am J Cardiol 2012;109:67-74.
60. Mohammed NA, Rafie I, Mahfouz A, Achkar K, Hajar R. Contrast-induced nephropathy. Heart Views 2013;14:106-16.
62. Mamoulakis C, Tsarouhas K, Fragkiadoulaki I, Heretis I, Wilks MF, et al. Contrast-induced nephropathy: basic concepts, pathophysiological implications and prevention strategies. Pharmacol Ther 2017;180:99-112.
64. Morcos R, Kucharik M, Bansal P, Al Taii H, Manam R, et al. Contrast-induced acute kidney injury: review and practical update. Clin Med Insights Cardiol 2019;13:1179546819878680.
65. Ward DB, Valentovic MA. Contrast induced acute kidney injury and direct cytotoxicity of iodinated radiocontrast media on renal proximal tubule cells. J Pharmacol Exp Ther 2019;370:160-71.
66. Celik T, Yilmaz MI, Balta S, Ozturk C, Unal HU, et al. The relationship between plasma whole blood viscosity and cardiovascular events in patients with chronic kidney disease. Clin Appl Thromb 2017;23:663-70.
67. Peters SAE, Woodward M, Rumley A, Tunstall-Pedoe HD, Lowe GDO. Plasma and blood viscosity in the prediction of cardiovascular disease and mortality in the Scottish Heart Health Extended Cohort Study. Eur J Prev Cardiol 2017;24:161-7.
68. Sugimori H, Tomoda F, Koike T, Kurosaki H, Masutani T, et al. Increased blood viscosity is associated with reduced renal function and elevated urinary albumin excretion in essential hypertensives without chronic kidney disease. Hypertens Res 2013;36:247-51.
69. Ferrario CM, Mullick AE. Renin angiotensin aldosterone inhibition in the treatment of cardiovascular disease. Pharmacol Res 2017;125:57-71.
70. Gavras I, Gavras H. Angiotensin II as a cardiovascular risk factor. J Hum Hypertens 2002;16 Suppl 2:S2-6.
71. Ibrahim NE, Shrestha S, McCarthy C, Lyass A, Li Y, et al. Endothelin-1 predicts incident heart failure, incident myocardial infarction, cardiovascular mortality, and all-cause mortality in patients undergoing diagnostic coronary angiography: results from the catheter sampled blood archive in cardiovascular disease (CASABLANCA) study. J Am Coll Cardiol 2018;71:A773.
72. Ozkok S, Ozkok A. Contrast-induced acute kidney injury: A review of practical points. World J Nephrol 2017;6:86-99.
73. Reiss AB, Grossfeld D, Kasselman LJ, Renna HA, Vernice NA, et al. Adenosine and the cardiovascular system. Am J Cardiovasc Drugs 2019;19:449-64.
74. Huang YT, Chen YY, Lai YH, Cheng CC, Lin TC, et al. Resveratrol alleviates the cytotoxicity induced by the radiocontrast agent, ioxitalamate, by reducing the production of reactive oxygen species in HK-2 human renal proximal tubule epithelial cells in vitro. Int J Mol Med 2016;37:83-91.
75. Jeong BY, Lee HY, Park CG, Kang J, Yu SL, et al. Oxidative stress caused by activation of NADPH oxidase 4 promotes contrast-induced acute kidney injury. PLoS One 2018;13:e0191034.
76. Chen Q, Zhang Y, Ding D, Xia M, Li D, et al. Estimated glomerular filtration rate and mortality among patients with coronary heart disease. PLoS One 2016;11:e0161599.
77. Dan Dunn J, Alvarez LAJ, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol 2015;6:472-85.
79. Plotnikov EY, Pevzner IB, Zorova LD, Chernikov VP, Prusov AN, et al. Mitochondrial damage and mitochondria-targeted antioxidant protection in LPS-induced acute kidney injury. Antioxidants 2019;8:176.
80. Tang C, Han H, Yan M, Zhu S, Liu J, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 2018;14:880-97.
81. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochim Biophys Acta - Mol Basis Dis 2017;1863:1066-77.
82. Nicolson GL. Mitochondrial dysfunction and chronic disease: Treatment with natural supplements. Integr Med (Encinitas) 2014;13:35-43.
83. Senoner T, Dichtl W. Oxidative stress in cardiovascular diseases: Still a therapeutic target? Nutrients 2019;11:2090.
84. Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko AV, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med 2018;50:121-7.
85. Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med 2019;51:1-13.
86. Siasos G, Tsigkou V, Kosmopoulos M, Theodosiadis D, Simantiris S, et al. Mitochondria and cardiovascular diseases - from pathophysiology to treatment. Ann Transl Med 2018;6:256.
87. Korge P, John SA, Calmettes G, Weiss JN. Reactive oxygen species production induced by pore opening in cardiac mitochondria: the role of complex II. J Biol Chem 2017;292:9896-905.
88. Manskikh VN, Gancharova OS, Nikiforova AI, Krasilshchikova MS, Shabalina IG, et al. Age-associated murine cardiac lesions are attenuated by the mitochondria-targeted antioxidant SkQ1. Histol Histopathol 2015;30:353-60.
89. Behringer EJ, Segal SS. Impact of aging on calcium signaling and membrane potential in endothelium of resistance arteries: a role for mitochondria. J Gerontol A Biol Sci Med Sci 2017;72:1627-37.
90. Bigelman E, Cohen L, Aharon-Hananel G, Levy R, Rozenbaum Z, et al. Pathological presentation of cardiac mitochondria in a rat model for chronic kidney disease. PLoS One 2018;13:e0198196.
91. Kocas C, Yildiz A, Abaci O, Karaca OS, Firdin N, et al. Platelet-to-lymphocyte ratio predicts contrast-induced nephropathy in patients with non-ST-segment elevation acute coronary syndrome. Angiology 2015;66:964-8.
92. Yuan Y, Qiu H, Hu X, Luo T, Gao X, et al. Predictive value of inflammatory factors on contrast-induced acute kidney injury in patients who underwent an emergency percutaneous coronary intervention. Clin Cardiol 2017;40:719-25.
93. Zhao K, Li Y, Gao Q. Role of red blood cell distribution width in predicting contrast induced nephropathy in patients with stable angina pectoris undergoing percutaneous coronary intervention. Int J Cardiol 2015;197:276-8.
94. Zorlu C, Koseoglu C. Comparison of the relationship between inflammatory markers and contrast-induced nephropathy in patients with acute coronary syndrome after coronary angiography. Angiology 2020;71:249-55.
95. Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal Syndrome. J Am Coll Cardiol 2008;52:1527-39.
96. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004;351:1296-305.
97. Schiffrin EL, Lipman ML, Mann JFE. Chronic kidney disease: effects on the cardiovascular system. Circulation 2007;116:85-97.
98. Tokuyama H, Kelly DJ, Zhang Y, Gow RM, Gilbert RE. Macrophage infiltration and cellular proliferation in the non-ischemic kidney and heart following prolonged unilateral renal ischemia. Nephron Physiol 2007;106:54-62.
99. Halliwell B. Role of free radicals in the neurodegenerative diseases: Therapeutic implications for antioxidant treatment. Drugs Aging 2001;18:685-716.
100. Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, et al. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol 2018;100:1-19.
101. Tang X, Luo YX, Chen HZ, Liu DP. Mitochondria, endothelial cell function, and vascular diseases. Front Physiol 2014;5:175.
102. Cobley J, Noble A, Bessell R, Guille M, Husi H. Reversible thiol oxidation inhibits the mitochondrial ATP synthase in xenopus laevis oocytes. Antioxidants 2020;9:215.
103. Cobley JN, Noble A, Jimenez-Fernandez E, Valdivia Moya MT, Guille M, et al. Catalyst-free Click PEGylation reveals substantial mitochondrial ATP synthase sub-unit alpha oxidation before and after fertilisation. Redox Biol 2019;26:101258.
104. Plotnikov EY, Zorov DB. Pros and cons of use of mitochondria-targeted antioxidants. Antioxidants 2019;8:316.
105. Liguori I, Russo G, Curcio F, Bulli G, Aran L, et al. Oxidative stress, aging, and diseases. Clin Interv Aging 2018;13:757-72.
106. Abete P, Napoli C, Santoro G, Ferrara N, Tritto I, et al. Age-related decrease in cardiac tolerance to oxidative stress. J Mol Cell Cardiol 1999;31:227-36.
107. Persson PB, Hansell P, Liss P. Pathophysiology of contrast medium-induced nephropathy. Kidney Int 2005;68:14-22.
108. Caiazza A, Russo L, Sabbatini M, Russo D. Hemodynamic and tubular changes induced by contrast media. Biomed Res Int 2014;2014:578974.
109. Brinkley TE, Nicklas BJ, Kanaya AM, Satterfield S, Lakatta EG, et al. Plasma oxidized low-density lipoprotein levels and arterial stiffness in older adults the health, aging, and body composition study. Hypertension 2009;53:846-52.
110. Zuliani G, Morieri ML, Volpato S, Vigna GB, Tch CB, et al. Determinants and clinical significance of plasma oxidized LDLs in older individuals. A 9 years follow-up study. Atherosclerosis 2013;226:201-7.
111. Burtenshaw D, Kitching M, Redmond EM, Megson IL, Cahill PA. Reactive Oxygen Species (ROS), Intimal Thickening, and Subclinical Atherosclerotic Disease. Front Cardiovasc Med 2019;6:89.
112. Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atheroscler Rep 2017;19:42.
113. Rivera J, Sobey CG, Walduck AK, Drummond GR. Nox isoforms in vascular pathophysiology: insights from transgenic and knockout mouse models. Redox Rep 2010;15:50-63.
114. Craige SM, Kant S, Reif M, Chen K, Pei Y, et al. Endothelial NADPH oxidase 4 protects ApoE-/- mice from atherosclerotic lesions. Free Radic Biol Med 2015;89:1-7.
115. Liu XH, Zhang QY, Pan LL, Liu SY, Xu P, et al. NADPH oxidase 4 contributes to connective tissue growth factor expression through Smad3-dependent signaling pathway. Free Radic Biol Med 2016;94:174-84.
116. Gray SP, Di Marco E, Kennedy K, Chew P, Okabe J, et al. Reactive oxygen species can provide atheroprotection via NOX4-dependent inhibition of inflammation and vascular remodeling. Arterioscler Thromb Vasc Biol 2016;36:295-307.
117. Wu RF, Ma Z, Liu Z, Terada LS. Nox4-derived H
118. Martin D, Li Y, Yang J, Wang G, Margariti A, et al. Unspliced X-box-binding protein 1 (XBP1) protects endothelial cells from oxidative stress through interaction with histone deacetylase 3. J Biol Chem 2014;289:30625-34.
119. Mocellin S, Bronte V, Nitti D. Nitric oxide, a double edged sword in cancer biology: searching for therapeutic opportunities. Med Res Rev 2007;27:317-52.
120. Lemkens P, Nelissen J, Meens MJPMT, Janssen BJA, Schiffers PMH, et al. Dual neural peptidase/endothelin-converting enzyme inhibition improves endothelial function in mesenteric resistance arteries of young spontaneously hypertensive rats. J Hypertens 2012;30:1799-808.
121. Barhoumi T, Briet M, Kasal DA, Fraulob-Aquino JC, Idris-Khodja N, et al. Erythropoietin-induced hypertension and vascular injury in mice overexpressing human endothelin-1: exercise attenuated hypertension, oxidative stress, inflammation and immune response. J Hypertens 2014;32:784-94.
122. Uchmanowicz I. Oxidative stress, frailty and cardiovascular diseases: current evidence. Adv Exp Med Biol 2020;1216:65-77.
123. McCullough PA, Adam A, Becker CR, Davidson C, Lameire N, et al. Risk prediction of contrast-induced nephropathy. Am J Cardiol 2006;98:27K-36.
124. Liu ZZ, Schmerbach K, Lu Y, Perlewitz A, Nikitina T, et al. Iodinated contrast media cause direct tubular cell damage, leading to oxidative stress, low nitric oxide, and impairment of tubuloglomerular feedback. Am J Physiol Renal Physiol 2014;306:F864-72.
125. Pisani A, Riccio E, Andreucci M, Faga T, Ashour M, et al. Role of reactive oxygen species in pathogenesis of radiocontrast-induced nephropathy. Biomed Res Int 2013;2013:868321.
126. Briguori C, Visconti G, Focaccio A, Airoldi F, Valgimigli M, et al. Renal insufficiency after contrast media administration trial II (REMEDIAL II): renalguard system in high-risk patients for contrast-induced acute kidney injury. Circulation 2011;124:1260-9.
127. Goldenberg I, Matetzky S. Nephropathy induced by contrast media: pathogenesis, risk factors and preventive strategies. CMAJ 2005;172:1461-71.
128. Briguori C, Donnarumma E, Quintavalle C, Fiore D, Condorelli G. Contrast-induced acute kidney injury: potential new strategies. Curr Opin Nephrol Hypertens 2015;24:145-53.
129. Rezaee MA, Mohammadpour AH, Imenshahidi M, Mahmoudi M, Sankian M, et al. Protective effect of erythropoietin on myocardial apoptosis in rats exposed to carbon monoxide. Life Sci 2016;148:118-24.
130. Gibson KR, Neilson IL, Barrett F, Winterburn TJ, Sharma S, et al. Evaluation of the antioxidant properties of N-acetylcysteine in human platelets: Prerequisite for bioconversion to glutathione for antioxidant and antiplatelet activity. J Cardiovasc Pharmacol 2009;54:319-26.
131. Andreucci M, Faga T, Pisani A, Sabbatini M, Michael A. Acute kidney injury by radiographic contrast media: pathogenesis and prevention. Biomed Res Int 2014;2014:362725.
132. Kwasa EA, Vinayak S, Armstrong R. The role of inflammation in contrast-induced nephropathy. Br J Radiol 2014;87:20130738.
133. Toso A, Leoncini M, Maioli M, Tropeano F, Di Vincenzo E, et al. Relationship between inflammation and benefits of early high-dose rosuvastatin on contrast-induced nephropathy in patients with acute coronary syndrome: the pathophysiological link in the PRATO-ACS study. JACC Cardiovasc Interv 2014;7:1421-9.
134. Kaya A, Kaya Y, Topçu S, Günaydın ZY, Kurt M, et al. Neutrophil-to-lymphocyte ratio predicts contrast-induced nephropathy in patients undergoing primary percutaneous coronary intervention. Angiology 2014;65:51-6.
135. El Sayed AA, Haylor JL, El Nahas AM, Salzano S, Morcos SK. Haemodynamic effects of water-soluble contrast media on the isolated perfused rat kidney. Br J Radiol 1991;64:435-9.
136. Limbruno U, Caterina R. Vasomotor effects of iodinated contrast media: just side effects? Curr Vasc Pharmacol 2003;1:321-8.
137. Aggarwal KP, Narula S, Kakkar M, Tandon C. Nephrolithiasis: molecular mechanism of renal stone formation and the critical role played by modulators. Biomed Res Int 2013;2013:292953.
138. Okoye O, Ojogwu L, Unuigbe E, Oviasu E. Frequency and risk factors of contrast-induced nephropathy after contrast procedures in a Nigerian tertiary centre. West Afr J Med 2013;32:19-25.
139. Ambrose JA, Bhullar AS. Inflammation and thrombosis in coronary atherosclerosis: pathophysiologic mechanisms and clinical correlations. Eur Med J 2019;4:71-8.
140. Hartman J, Frishman WH. Inflammation and atherosclerosis: a review of the role of interleukin-6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiol Rev 2014;22:147-51.
141. Satilmis S, Karabulut A. Value of C-reactive protein/albumin ratio in predicting the development of contrast-induced nephropathy in patients with non-ST elevation myocardial infarction. Angiology 2020;71:366-71.
142. Buyuklu M, Kandemir F, Ozkaraca M, Set T, Bakirci EM, et al. Benefical effects of lycopene against contrast medium-induced oxidative stress, inflammation, autophagy, and apoptosis in rat kidney. Hum Exp Toxicol 2015;34:487-96.
143. Buyuklu M, Mehmet Kandemir F, Ozkaraca M, Set T, Murat Bakirci E, et al. Protective effect of curcumin against contrast induced nephropathy in rat kidney: what is happening to oxidative stress, inflammation, autophagy and apoptosis? Eur Rev Med Pharmacol Sci 2014;18:461-70.
144. Greaves DR, Channon KM. Inflammation and immune responses in atherosclerosis. Trends Immunol 2002;23:535-41.
145. Ruparelia N, Chai JT, Fisher EA, Choudhury RP. Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nat Rev Cardiol 2017;14:133-44.
146. Ong SB, Hernández-Reséndiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, et al. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther 2018;186:73-87.
147. Rajendran K, Devarajan N, Ganesan M, Ragunathan M. Obesity, inflammation and acute myocardial infarction - expression of leptin, IL-6 and high sensitivity-CRP in Chennai based population. Thromb J 2012;10:13.
148. Senguttuvan NB, Subramanian A, Agarwal G, Mishra S, Bahl VK. Association of Cytokines IL6, IL10, IL18, TNFα in acute coronary syndrome. J Cardiol Vasc Med 2019;5:1-9.
149. Maekawa N, Wada H, Kanda T, Niwa T, Yamada Y, et al. Improved myocardial ischemia/reperfusion injury in mice lacking tumor necrosis factor-α. J Am Coll Cardiol 2002;39:1229-35.
150. Riad A, Jäger S, Sobirey M, Escher F, Yaulema-Riss A, et al. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J Immunol 2008;180:6954-61.
151. Satoh M, Shimoda Y, Maesawa C, Akatsu T, Ishikawa Y, et al. Activated toll-like receptor 4 in monocytes is associated with heart failure after acute myocardial infarction. Int J Cardiol 2006;109:226-34.
152. Shishido T, Nozaki N, Yamaguchi S, Shibata Y, Nitobe J, et al. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation 2003;108:2905-10.
153. Schlitt A, Heine GH, Blankenberg S, Espinola-Klein C, Dopheide JF, et al. CD14+CD16+ monocytes in coronary artery disease and their relationship to serum TNF-α levels. Thromb Haemost 2004;92:419-24.
154. Mishra J, Dent C, Tarabishi R, Mitsnefes MM, Ma Q, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 2005;365:1231-8.
155. Bachorzewska-Gajewska H, Malyszko J, Sitniewska E, Malyszko JS, Dobrzycki S. Neutrophil-gelatinase-associated lipocalin and renal function after percutaneous coronary interventions. Am J Nephrol 2006;26:287-92.
156. Malyszko J, Malyszko JS, Bachorzewska-Gajewska H, Poniatowski B, Dobrzycki S, et al. Neutrophil gelatinase-associated lipocalin is a new and sensitive marker of kidney function in chronic kidney disease patients and renal allograft recipients. Transplant Proc 2009;41:158-61.
157. Haase M, Devarajan P, Haase-Fielitz A, Bellomo R, Cruz DN, et al. The outcome of neutrophil gelatinase-associated lipocalin-positive subclinical acute kidney injury. J Am Coll Cardiol 2011;57:1752-61.
158. Zappitelli M, Washburn KK, Arikan AA, Loftis L, Ma Q, et al. Urine neutrophil gelatinase-associated lipocalin is an early marker of acute kidney injury in critically ill children: a prospective cohort study. Crit Care 2007;11:R84.
159. Nakamura T, Sugaya T, Node K, Ueda Y, Koide H. Urinary excretion of liver-type fatty acid-binding protein in contrast medium-induced nephropathy. Am J Kidney Dis 2006;47:439-44.
160. Baramova EN, Bajou K, Remacle A, L’hoir C, Krell HW, et al. Involvement of PA/plasmin system in the processing of pro-MMP-9 and in the second step of pro-MMP-2 activation. FEBS Lett 1997;405:157-62.
161. Stringer KA, Bose SK, McCord JM. Antiinflammatory activity of tissue plasminogen activator in the carrageenan rat footpad model. Free Radic Biol Med 1997;22:985-8.
162. Yu LR, Sun J, Daniels JR, Cao Z, Schnackenberg L, et al. Aptamer-based proteomics identifies mortality-associated serum biomarkers in dialysis-dependent AKI patients. Kidney Int Reports 2018;3:1202-13.
163. Roelofs JJTH, Rouschop KMA, Leemans JC, Claessen N, de Boer AM, et al. Tissue-type plasminogen activator modulates inflammatory responses and renal function in ischemia reperfusion injury. J Am Soc Nephrol 2006;17:131-40.
164. Hu K, Yang J, Tanaka S, Gonias SL, Mars WM, et al. Tissue-type plasminogen activator acts as a cytokine that triggers intracellular signal transduction and induces matrix metalloproteinase-9 gene expression. J Biol Chem 2006;281:2120-7.
165. Lin L, Wu C, Hu K. Tissue plasminogen activator activates NF-κB through a pathway involving annexin A2/CD11b and integrin-linked kinase. J Am Soc Nephrol 2012;23:1329-38.
166. Lin L, Jin Y, Mars WM, Reeves WB, Hu K. Myeloid-derived tissue-type plasminogen activator promotes macrophage motility through FAK, Rac1, and NF-κB pathways. Am J Pathol 2014;184:2757-67.
167. Ferguson-Smith AC, Chen YF, Newman MS, May LT, Sehgal PB, et al. Regional localization of the interferon- β2 B-cell stimulatory factor 2/hepatocyte stimulating factor gene to human chromosome 7p15-p21. Genomics 1988;2:203-8.
168. Ling W, Zhaohui N, Ben H, Leyi G, Jianping L, et al. Urinary IL-18 and NGAL as early predictive biomarkers in contrast-induced nephropathy after coronary angiography. Nephron - Clin Pract 2008;108:c176-81.
169. Nechemia-Arbely Y, Barkan D, Pizov G, Shriki A, Rose-John S, et al. IL-6/IL-6R axis plays a critical role in acute kidney injury. J Am Soc Nephrol 2008;19:1106-15.
170. Chen J, Hartono JR, John R, Bennett M, Zhou XJ, et al. Early interleukin 6 production by leukocytes during ischemic acute kidney injury is regulated by TLR4. Kidney Int 2011;80:504-15.
171. Alladina JW, Levy SD, Hibbert KA, Januzzi JL, Harris RS, et al. Plasma concentrations of soluble suppression of tumorigenicity-2 and interleukin-6 are predictive of successful liberation from mechanical ventilation in patients with the acute respiratory distress syndrome. Crit Care Med 2016;44:1735-43.
172. Nogare AL, Dalpiaz T, Veronese FJ, Gonçalves LF, Manfro RC. Noninvasive analyses of kidney injury molecule-1 messenger RNA in kidney transplant recipients with graft dysfunction. Transplant Proc 2012;44:2297-9.
173. Slocum JL, Heung M, Pennathur S. Marking renal injury: can we move beyond serum creatinine? Transl Res 2012;159:277-89.
174. Soto K, Coelho S, Rodrigues B, Martins H, Frade F, et al. Cystatin C as a marker of acute kidney injury in the emergency department. Clin J Am Soc Nephrol 2010;5:1745-54.
175. Marenzi G. Can contrast-induced nephropathy after percutaneous coronary intervention be accurately predicted with a risk score? Nat Clin Pract Cardiovasc Med 2005;2:80-1.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Cervantes-Gracia K, Raja K, Llanas-Cornejo D, Cobley JN, Megson IL, Chahwan R, Husi H. Oxidative stress and inflammation in the development of cardiovascular disease and contrast induced nephropathy. Vessel Plus 2020;4:27. http://dx.doi.org/10.20517/2574-1209.2020.22
AMA Style
Cervantes-Gracia K, Raja K, Llanas-Cornejo D, Cobley JN, Megson IL, Chahwan R, Husi H. Oxidative stress and inflammation in the development of cardiovascular disease and contrast induced nephropathy. Vessel Plus. 2020; 4: 27. http://dx.doi.org/10.20517/2574-1209.2020.22
Chicago/Turabian Style
Cervantes-Gracia, Karla, Khuram Raja, Daniel Llanas-Cornejo, James N. Cobley, Ian L. Megson, Richard Chahwan, Holger Husi. 2020. "Oxidative stress and inflammation in the development of cardiovascular disease and contrast induced nephropathy" Vessel Plus. 4: 27. http://dx.doi.org/10.20517/2574-1209.2020.22
ACS Style
Cervantes-Gracia, K.; Raja K.; Llanas-Cornejo D.; Cobley JN.; Megson IL.; Chahwan R.; Husi H. Oxidative stress and inflammation in the development of cardiovascular disease and contrast induced nephropathy. Vessel Plus. 2020, 4, 27. http://dx.doi.org/10.20517/2574-1209.2020.22
About This Article
Copyright

Data & Comments
Data


 Cite This Article 38 clicks
Cite This Article 38 clicks

 Like This Article 23
likes
Like This Article 23
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.