
Features of cholesterol metabolism in macrophages in immunoinflammatory diseases
 0
0 , ...
, ... Abstract
Immune-inflammatory rheumatological diseases are a large group of pathological conditions that lead to chronic inflammation and organ damage. Many autoimmune diseases are associated with a high risk of cardiovascular complications, including atherosclerosis. Inflammation plays a significant role in the development and accelerated course of atherosclerotic lesions. Disorders of lipid metabolism are closely associated with the functions of cells of the immune system and can contribute to the development of these diseases. Cholesterol and lipids are involved in various cellular processes, including intercellular recognition, signal transmission and energy supply. The effect of cholesterol metabolism on the immune response is of great importance and is being actively investigated. Further study of the mechanism of cholesterol efflux from cells may be the key to understanding the relationship between immune-inflammatory and cardiovascular diseases. In this review, we have summarized data on cholesterol metabolism and its effect on the development of pathological conditions.
Keywords
INTRODUCTION
Rheumatological diseases are a group of autoimmune disorders that occur chronically and affect many organs or organ systems, leading to a high risk of mortality and disability. The estimated prevalence in developed countries ranges from 3%-5%, according to the various evaluations in different populations. In general, if we evaluate the entire population of the globe, then rheumatological diseases are detected in 1% of people[1]. The most common diseases of this group are systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), systemic scleroderma, idiopathic inflammatory myopathy and Sjogren’s syndrome[2].
Recently, the connection of the immune system with metabolic processes in autoimmune and rheumatological diseases has been widely studied. The research focuses on key metabolic pathways, which comprise the pentose phosphate pathway, glycolysis, the tricarboxylic acid cycle, amino acid metabolism, oxidation and synthesis of fatty acids (FAs). Studies of lipid metabolism have been conducted for many years[3]. An altered lipid profile is often found in rheumatological diseases. Dyslipidemia is a generally accepted risk factor for the development of atherosclerosis and rheumatic diseases[2,4]. Its feature is a low level of high-density lipoproteins (HDL), high levels of low-density lipoproteins (LDL), triglycerides (TG) and total cholesterol (TC). The increased risk of atherosclerosis and damage to target organs, such as the central nervous system and kidneys, depend on high levels of LDL and/or abnormal levels of HDL in blood plasma[5,6].
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease, the prevalence of which is approximately 0.5%-1% of the population. In this disease, a common cause of death is cardiovascular complications[7]. Most of the data from genetic analysis, tissue analysis, animal models and clinical studies indicate an immuno-mediated etiology that contributes to chronic inflammation and joint destruction[8].
The development of RA and cardiovascular diseases (CVD) is influenced by common inflammatory mediators, posttranslational modifications of proteins, immune responses, changes in the composition and function of lipoproteins (LPS), oxidative stress and endothelial dysfunction[9,10]. The role of the central immune system in the development of CVD is being proven by more and more studies. Proinflammatory cytokines that increase atherogenesis are involved in the development of RA[11]. Understanding the unique mechanisms of CVD development in RA will help identify new goals to reduce significantly cardiovascular (CV) risk in patients[9,12].
SLE is a chronic autoimmune disease that affects mainly women of reproductive age. At the same time, the human immune system makes a mistake and produces autoantibodies. The autoantibodies mistakenly identify the body’s own cells as being foreign and attack them. The result is an autoimmune reaction, circulating immune complexes are deposited in various organs, which leads to inflammation[13]. Deviations of the immune system, as well as hereditary, hormonal and environmental factors, affect the manifestation of organ damage. The genetic contribution to the progression of the disease is undeniable, but the etiology is still unclear. Gene polymorphisms that contribute to the development of SLE include single nucleotide polymorphisms, gene defects, duplications, and aberrant expression of splicing variants[14]. Patients with SLE develop atherosclerosis faster than people without this pathology. It is necessary to establish a link between cardiovascular diseases and SLE[15,16]. Hypertension, dyslipidemia and elevated levels of oxidized lipids, the presence of a large number of autoantibodies and inflammation can contribute to the progression of atherosclerosis in patients with SLE[17].
In general, atherosclerosis is a common chronic inflammatory disease in which damage to large vessels develops. Concomitant pathologies can be coronary heart disease (CHD), stroke and peripheral vascular diseases[18]. With atherosclerosis, cholesterol accumulates in the intima space of the vessels, because there is an increase in the amount and difficulty in the outflow of LDL[19]. The remaining LDLs are modified and engulfed by phagocytes[20]. This process leads to the formation of plaques, which consist of monocytes and cholesterol. There is a strong association between cholesterol, LDL and apolipoprotein, including apolipoprotein B (ApoB), and atherosclerosis, which makes them the main target for research[21].
CHOLESTEROL METABOLISM IN CELLS, TRANSPORT AND DISTRIBUTION AT THE INTRACELLULAR LEVEL, CHOLESTEROL OUTFLOW PATHWAYS, THE ROLE OF MACROPHAGES
The classical theory of the mechanism of atherosclerosis development presented it as a disease with impaired lipid metabolism. It was subsequently changed due to more in-depth studies[22,23]. Now it is impossible to deny the role of inflammatory pathways in the spread of atherosclerosis and the occurrence of acute coronary syndrome[11]. In many chronic inflammatory and autoimmune diseases, there is an increased risk of CVD. It remains to be seen whether the mediators of atherogenesis are common to all chronic inflammatory pathologies[22]. In any case, in the development of this pathology, an important role is played by the disorder of cholesterol metabolism. Multiple evidence supports that enhanced cholesterol efflux from foam cells by HDL particles is a promising antiatherogenic strategy[24].
Reverse cholesterol transport (RCT) is the process of transferring excess cholesterol from peripheral tissues to plasma using HDL. After that, it enters the liver, from where it is removed along with bile or metabolized before excretion [Figure 1][24]. Cholesterol must be in a non-esterified form so that it can be eliminated from the cells. This process was found in experiments in vitro and in vivo, where the hydrolysis of lipid droplets in foam cells limits the RCT rate[25]. Free cholesterol is released from lipid droplets by hydrolysis of cholesterol ester. After that, it can move to the plasma membrane and pass to the cholesterol acceptor, or be esterified again by cholesterol acyltransferase[26].

Figure 1. Reverse cholesterol transport. ABCA1: ATP binding cassette transporter A1; ABCG1: ATP binding cassette transporter G1; ABCG5: ATP binding cassette transporter G5; ABCG8: ATP binding cassette transporter G8; ApoA-I: apolipoprotein A-I; ApoB: apolipoprotein B; CE: cholesterol ester; CEPT: cholesteryl ester transfer protein; EL: endothelial lipase; HDL: high density lipoproteins; LCAT: lecithin-cholesterol acyltransferase; LDL: low-density lipoproteins; LDLR: low-density lipoprotein receptor; LXR: liver X receptor; PLTP: phospholipid-transfer protein; SR-A: scavenger receptor class A; SR-B1: scavenger receptor class B type 1; TG: triglycerides; VLDL: very-low-density lipoprotein.
A decrease or increase in the production of various enzymes affects the metabolism of cholesterol in cells. In particular, liver X receptors (LXRs) are the key sterol-sensitive transcription factors in macrophages that control the intracellular balance of cholesterol and lipids[27]. LXRs regulate the expression of numerous efflux pathway genes, including the ATP-binding cassette (ABC) proteins ABCA1 (member 1 of human transporter sub-family ABCA) and ABCG1 (ATP-binding cassette sub-family G member 1), which are the main carriers of cellular cholesterol from foam cells[28]. Foam cells are cholesterol-loaded macrophages and are the main link in the pathogenesis of atherosclerosis[29]. ABCA1 serves for the outflow of free cholesterol into apolipoprotein AI (ApoA-I) and is important for HDL biogenesis[30], and ABCG1 promotes the transfer of cholesterol to the plasma membrane from the endoplasmic reticulum (ER)[31]. After the transfer of cholesterol to HDL particles, they are esterified by lecithin-cholesterol acyltransferase (LCAT) to form a CE, which leads to the formation of mature HDLs. Plasma phospholipid-transfer protein (PLTP) and cholesteryl ester transfer protein (CETP) both play a major role in the metabolism of those lipoproteins. CEPT mediates the transport of TG and CE between HDL and non-HDL particles, and PLTP can stabilize the activity of ABCA1. Cholesterol enters macrophages as a result of the entry of LPs into the cell or as a result of efferocytosis of apoptotic cells[24]. Pre-β-HDL, which are formed due to the communication of ABCA1 and monomolecular apoA-I, are effective acceptors of free cholesterol transported from the plasma membrane of peripheral cells[32]. Excess cholesterol is removed from cells to extracellular acceptors or converted into CE and stored in cells in the form of cytosolic lipid droplets[24]. HDL containing cholesterol esters bind to SR-B1 (scavenger receptor class B type 1), which contributes to the efflux of cholesterol back into the liver[33]. Another protective reaction to an increased level of cellular cholesterol is the inhibition of SREBP (sterol regulatory element-binding protein) processing, which leads to a decrease in the expression of the gene for the enzyme HMGCR (3-hydroxy-3-methyl-glutaryl-coenzyme A reductase) - the main target of statins, which limits the rate of cholesterol production and is under the influence of the mechanism of negative feedback[24]. LXRs contribute to the removal of excess cholesterol in response to its elevated levels in cells[34]. SREBP2 protein promotes the biosynthesis and absorption of cholesterol if its content in cells decreases[35]. Actually, SREBP2 is the main transcription factor for the low-density lipoprotein receptor (LDLR). When cholesterol levels are low, SREBP2 is activated and triggers the transcription of genes encoding proteins that affect cholesterol metabolism[36]. SCAP (SREBP cleavage-activating protein) is a sterol-regulated companion protein that delivers SREBP from the site of their synthesis in the ER to the site of their cleavage in the Golgi complex[37]. There it is cleaved by two serine proteases (S1P and S2P), releasing the cytosolic NH2-terminal domains of the transcription factor[38]. These fragments penetrate into the nucleus, where they combine with steroid regulatory elements in the enhancer regions of more than twenty genes, resulting in the synthesis of cholesterol and unsaturated FAs[39]. In this case, the LXRs remain in a repressive state. Under high cholesterol conditions, a trimolecular complex consisting of the INSIG (insulin-induced gene), SCAP and SREBP2, remains in the ER, blocking SREBP2 activation and cholesterol synthesis[40].
Macrophages metabolize cholesterol. A certain amount of cholesterol remains in the cytosolic droplets of CE, then the excess again passes into the bloodstream to be processed by the liver. The outflow of cholesterol from cells involves several mechanisms. The first of them triggers the metabolism of cellular cholesterol by direct interaction of HDL and ABC on the surface of the plasma membrane. To combine cholesterol with HDL, ABC transporters transfer phospholipids and cholesterol to the outer layer of the plasma membrane. The expression of ABC transporters is controlled by LXR transcription factors. The second mechanism of cholesterol efflux from macrophages can be mediated by the release of particles with cholesterol, which are called “microparticles”, “microdomains of cholesterol” or “exosomes”[41]. Scientists have hypothesized that a significant portion of cholesterol is associated with microparticles that are generated from the plasma membrane, but it is unclear how they were released[42]. Numerous investigations of cholesterol efflux by macrophages have concentrated on the ability of ABC transporters to transfer cellular cholesterol into LP particles. Cultured macrophages deliver cholesterol from the plasma membrane in the shape of “cholesterol microdomains”. These microdomains help to remove excess cholesterol[43]. Later it was established that cholesterol microdomains are not vesicles, but are branching structures of irregular shape originating from the plasma membrane. However, the way they exit the cell is not specified[44]. It has been shown that cultured macrophages secrete vesicular particles enriched with “available cholesterol” (cholesterol pool) from the plasma membrane into the surrounding substrate[45]. The ability of macrophages to secrete vesicles containing cholesterol increased the likelihood of cholesterol transfer to neighboring cells[46]. Later it was found that ABCA1-deficient macrophages carry cholesterol to neighboring smooth muscle cells (SMCs). Most of the cholesterol transported to SMCs was taken from an available pool in the plasma membrane of macrophages; therefore, it is possible that pools isolated by sphingolipids or phospholipids contribute to the movement of cholesterol[47]. Cholesterol moves from macrophages to SMCs through membrane junctions between cells. It is known that these structures are detected in cultured macrophages[48].
Another pathway for cholesterol transport is due to ORP (oxysterol-binding protein-related proteins) connected to OSBP (oxysterol-binding protein). They are a family of lipid binding/transferring proteins that can promote non-vesicular cholesterol transfer between lipid bilayers, improving cholesterol transport between subcellular membrane organelles[49]. ORP6 controls cholesterol efflux and HDL balance and may be a new regulator of the RCT pathway. Transcription of the ORP6 gene is regulated by LXR transcription factors, which turn on at high levels of cellular cholesterol and regulate the expression of genes involved in cholesterol metabolism[50].
In studies on models of macrophages without LXR, an increase in lesions occurred[51], and LXR agonists had the opposite effect[52]. Lipids play an important role in the polarization of macrophages. Experiments on mouse models have demonstrated that the effect on lipid metabolism in macrophages can improve the course of atherosclerosis. Overexpression of LXR in macrophages has an antiatherogenic effect due to an increase in cholesterol efflux[53]. However, when LXR is activated, side effects such as lipogenesis and hypertriglyceridemia are detected, so there is a need for new studies of other ligands[54]. It has been demonstrated that people with a specific variant of perilipin-2, a protein associated with lipid droplets, are less vulnerable to the onset and progression of atherosclerosis[55]. The mechanism of this protection includes the activation of LXR in primary macrophages originating from monocytes. Innate immunity has also become a potential tool for combating atherosclerosis[56].
FEATURES OF CHOLESTEROL METABOLISM IN MACROPHAGES IN RHEUMATOID ARTHRITIS, DISTURBANCE OF LIPID METABOLISM, CAUSES, CONSEQUENCES
Patients with RA are 2-3 times more likely to have atherosclerosis[57]. The mechanisms by which the development of atherosclerotic lesions in RA is accelerated are unknown. Systemic inflammation is believed to play a key role in this process[58]. According to this hypothesis, circulating levels of monocytes and platelets are elevated in patients with RA[58,59]. The role of monocytes in joint damage has been studied for a long time. There have been numerous studies on the link between myeloid cells and atherosclerosis[60].
Monocytes, neutrophils and platelets originate from the bone marrow (BM) by myelopoiesis from hematopoietic stem cells and progenitor cells (HSPCs). Accumulation of cellular cholesterol leads to hyperproliferation and increased myelopoiesis. HSPCs and myeloid progenitor cells regulate cholesterol efflux, as described above, through the ATP-binding cassette transporters ABCA1 and ABCG1 and cell surface apolipoprotein-E (apoE) [Figure 2][61]. In patients with RA, cholesterol metabolism is disrupted owing to the inhibition of the expression of ABCA1 and ABCG1[62]. Proteomic analysis in patients with RA showed the presence of acute phase proteins (SAA - serum amyloid A) and complement factors (B, C3, C9)[63]. There is also a reduced level of HDL, an increase in the amount of proinflammatory and oxidized lipids, a violation of antioxidant activity[64]. Elevated levels of ox-LDL were detected in synovial fluid and synovial membrane and positively associated with CVD in patients with RA[65]. These damages in cholesterol transport can affect the cellular balance of cholesterol and lead to an increased risk of CVD[66].
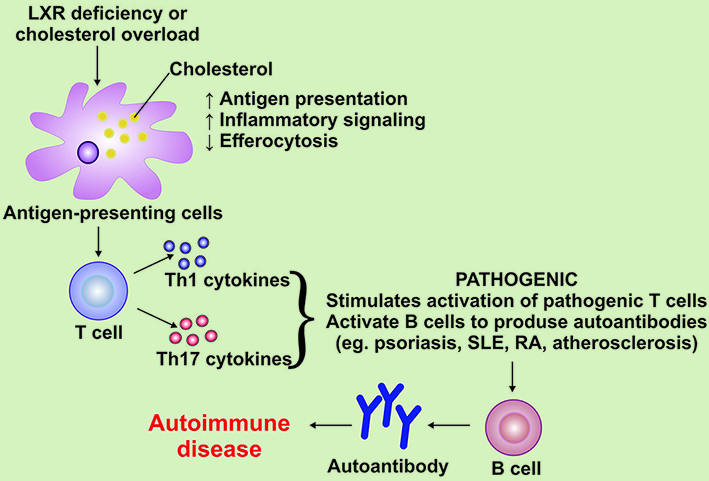
Figure 2. The effect of cholesterol metabolism on the progression of autoimmune diseases. LXR: Liver X receptor; RA: rheumatoid arthritis; SLE: systemic lupus erythematosus; Th1: T helper 1 cells; Th17: T helper 17 cells.
Inflammatory arthritis has been found to impair the regression of atherosclerotic lesions and accelerate atherogenesis in preclinical models of RA[67]. Through experiments in mouse models of RA, it has become known that bone marrow-derived HSPCs have impaired cellular balance of cholesterol, possibly because of systemic inflammation. Myeloid cells retain this damage in cholesterol transport, which may contribute to the appearance of foam cells and increase the incidence of CV complications in RA[68].
The team of scientists found that the activity of myeloperoxidase (MPO) in blood plasma was greatly increased in patients with RA compared to healthy controls in terms of age, ethnicity and gender[69]. MPO is a protein found in the granules of neutrophils and monocytes that generates reactive oxygen species to destroy invading pathogens[70]. MPO levels are also elevated in atherosclerotic lesions[71] and are related to the prevalence of coronary heart disease in the overall population[72]. ApoA-I is a selective target for nitration and chlorination catalyzed by MPO. Oxidation of HDL and apoA-I leads to selective suppression of ABCA1, which impairs cholesterol efflux[73].
In patients with RA, dyslipidemia can be stopped without the use of statins, using anti-inflammatory and anti-rheumatic drugs[74]. Glucocorticoids have both atheroprotective and proatherogenic effects. The reason for these differences is the ability of glucocorticoids to act on several types of cells in the vasculature, with different effects depending on the concentration and target cells[75].
LIPID METABOLISM DISTURBANCES IN SYSTEMIC LUPUS ERYTHEMATOSUS
CHD and atherosclerosis are more common in patients with SLE compared to the control group, which could not be predicted only by the main risk factors[76]. Since CVD deaths account for more than a third of all deaths in patients with SLE, it is obvious that CV complications are one of the main problems for patients with SLE[77].
HDL exhibit antioxidant, anti-inflammatory, antithrombotic, and antiapoptotic properties independent of cholesterol mobilization[78]. Patients with SLE with rapidly developing atherosclerosis have the progression of dysfunction and a decrease in HDL levels [Figure 2][79]. These data suggest that HDL is a target for reducing the frequency of CVD in patients with SLE[80].
Elimination of cholesterol from vascular macrophages is an important process to protect against atherosclerosis and improve CVD outcomes[81]. This process is mediated by OTC and allows the transfer of excess cholesterol and other lipids from macrophages in atherosclerotic lesions to the liver for excretion. The first and very important stage in RCT is the efflux of cholesterol from macrophages through HDL[80]. A violation of lipid metabolism is observed in the early diagnosis of SLE[82]. Numerous studies suggest that the reason for the decrease in the ability of cholesterol efflux in SLE is an elevated level of the SAA protein, which contributes to HDL dysfunction and reduces the ability to delete cholesterol from macrophages and move CE to the liver[83].
HDL oxidation may also help reduce cholesterol efflux in SLE. In HDL containing oxidized apoA-I, the capacity to drain cholesterol is reduced[84]. When a single methionine residue (Met-148) is oxidized in apoA-I, HDL loses the capacity to interact with LCAT, the enzyme responsible for the formation of CE, which is the main step in RCT. Thus, the oxidation of apoA-I may be one of the causes of impaired cholesterol efflux in SLE[85].
HDL can directly suppress inflammatory processes leading to the progression of atherosclerosis[86]. HDL inhibits TLR-induced generation of proinflammatory cytokines by macrophages at the transcriptional level. Activated transcriptional repressor (ATF3) of innate immune response genes moves to the nucleus and inhibits the stimulation of TLR-induced inflammatory cytokines[87]. HDL can also reduce the production of cell adhesion molecules activated by nuclear factor-κB (NF-κB), thereby preventing atherosclerotic disease[88]. HDL in healthy individuals contribute to the anti-inflammatory response. Lipoproteins derived from SLE patients induce a proinflammatory response. Scientists have found that HDL in SLE cannot suppress TLR-mediated cytokine induction[89]. It has also been found that HDL in SLE patients can activate PDGFRβ (platelet- derived growth factor receptor β) and increase chemotaxis and release of TNF-α[90].
The oxidation of HDL in SLE contributes to their binding to LOX1R (a lectin-like receptor of oxidized low-density lipoproteins 1), preventing nuclear translocation of ATF3 and leading to an enhanced synthesis of inflammatory cytokines[91,92]. Elevated oxLDL levels improve the adhesion of monocytes to activated endothelial cells by enhancing the expression of adhesion molecules and proinflammatory cytokines[92,93]. Then monocytes migrate to the intima of the arteries, absorbing oxLDL and foam cells are formed[87]. The use of statins appears to benefit patients with SLE, but further studies are needed to see a sustained positive effect[94].
BRIEF DESCRIPTION OF LIPID METABOLISM IN OTHER RHEUMATOLOGICAL DISEASES
Rheumatological diseases include multiple sclerosis (MS), a chronic progressive inflammatory and degenerative pathology of the central nervous system caused by autoimmune and inflammatory processes leading to demyelination and degeneration of neurons[95]. Proinflammatory lipids are the cause of the typical clinical symptoms related to many autoimmune rheumatic diseases (ARDs)[96]. Lipid metabolism affects many functions of immune cells[97]. The link between lowering cholesterol levels and the progression of MS has been confirmed[98]. In patients with MS with excessive accumulation of cholesterol or deficiency of LXR, there is a violation of the functions of T cells, which leads to the activation of B cells and the production of autoantibodies [Figure 2][99].
Psoriasis is a widespread skin disease affecting about 1%-3% of the general population[100]. Patients with psoriasis are more susceptible to obesity, dyslipidemia, atherosclerosis and non-alcoholic fatty liver disease. Researches show that lipid metabolism disorders are more common in these patients[101]. An important feature is that atherosclerotic plaques are similar to psoriatic ones. Both plaques develop due to chronic inflammatory conditions and are associated with immune processes that involve cytokines, T-lymphocytes, and thrombotic agents[102]. The analysis of research into lipid metabolism has shown that the levels of total cholesterol, LDL and very-low-density lipoproteins (VLDL) are higher in patients with psoriasis than in control groups[103]. The lipid profile of the patients showed a high level of lipids with Apo-B [Figure 3]. The amount of HDL does not change[104]. The antioxidant activity of HDL mediated by paraoxonase 1 (PON1) seems to persist in patients with psoriasis, despite the change in composition towards proinflammatory HDL particles. Since such a shift was associated with a violation of ABCA1-CEC, these results may be evidence of a link between psoriasis and CVD[105].

Figure 3. Cholesterol efflux defects in immunoinflammatory diseases. ABCA1-CEC: ABCA1-specific cellular cholesterol efflux capacity; ABCG1-CEC: ABGA1-specific cellular cholesterol efflux capacity; apoA-I: apolipoprotein A-I; apoB-I: apolipoprotein B-I; HDL: high density lipoproteins; PON-I: paraoxonase-I; SAA: serum amyloid A.
FEATURES OF CHOLESTEROL METABOLISM IN MACROPHAGES IN ATHEROSCLEROSIS AT DIFFERENT STAGES OF THE DISEASE. THE ROLE OF MACROPHAGES IN INITIATION AND PROGRESSION OF LESION
Studies of the initial stages of atherogenesis in human and animal models show that the main initial stage is the subendothelial accumulation of lipoproteins with apolipoprotein B (apoB-LPs)[106]. ApoB-LPs generated by the liver cells are secreted as VLDL, which are converted into atherogenic LDL in the bloodstream[107]. Intestinal apoB-LPs are formed in the form of chylomicrons, which turn into atherogenic particles due to lipolysis[108].
The first reaction to apoB-LPs is the activation of overlying endothelial cells, which leads to the recruitment of blood monocytes. Activated endothelial cells secrete chemokines that react with the corresponding chemokine receptor on monocytes and contribute to directed relocation[109]. Monocytes are formed from progenitor cells in the BM. The onset of monocyte growth is controlled by the content of cellular cholesterol, so it can affect atherogenesis. Mice whose monocyte progenitor cells have a defect in cholesterol efflux owing to a shortage of ABCA1 and ABCG1 transporters demonstrate an increase in the number of circulating monocytes (monocytosis) and the development of atherosclerosis[110]. After monocytes are firmly attached to the affected endothelial cells owing to the interplay of monocyte integrins with endothelial cell ligands[111,112]. Strong adhesion is accompanied by their penetration into the subendothelial space[113,114].
Mouse monocytes in early atheromas become macrophage-like and/or dendrite-like cells under the action of macrophage-colony stimulating factor (M-CSF)[115,116]. Even at the initial stages, macrophages and dendritic cells contain lipid droplets linked to the cytoplasmic membrane (foam cells)[117,118]. Further formation of foam cells starts when phagocytes engulf and process apoB-LPs[119]. CE are then hydrolyzed in endosomes to FC and FA[120]. FC delivery to the ER plays an important role in downregulating LDL receptors and endogenous cholesterol synthesis by suppressing the SREBP[121]. FC transported from lysosomes and CE from non-hydrolyzed droplets can penetrate the plasma membrane and be accessible for efflux from the cell[119]. Defective transfer of FC from lysosomes in damaged macrophages creates a barrier to cholesterol efflux and damage regression[122]. Once on the plasma membrane, cholesterol is transported to its outer layer, where it is removed from cells by ABCA1- and ABCG1-mediated transport to apoA-I and HDL or by “passive diffusion” to low-cholesterol HDL[123,124].
In experimental models, the classic inflammatory phenotype of macrophages has been termed M1 and is often activated by in vitro incubation of macrophages with a combination of IFN-γ and LPS. A subpopulation of M2 macrophages has also been detected in vitro. The M2 phenotype can be activated by adding IL-4 and IL-13, which suppress the M1 phenotype and promote the production of IL-10 and TGF-β (transforming growth factor beta)[125]. There are much more subpopulations of macrophages, since macrophages encounter various microenvironments, the signals of which influence them[126]. Studies in Apoe-/- mice have demonstrated that M2 macrophages colonize initial fatty streaks. The further the lesion progresses, the more common the M1 phenotype of macrophages is[127].
Stages of atherosclerotic changes in the vascular wall. At the stage of initiation of an atherosclerotic lesion, most monocytes differentiate into macrophages. This process is crucial for the further resolution of lesions[128]. Macrophages congregate in receptive areas of the arteries due to the expression of endothelial adhesion molecules and the presence of apoB-LPs in the subendothelium[125]. Chemokines produced by endothelial cells and macrophages attract even more monocytes[129,130]. Macrophages absorb modified lipids and other substances from the subendothelium. The fatty streak with macrophages increases due to the accumulation of a large number of macrophages and their transformation into foam cells[131]. Early lesions may resolve with efferocytosis[125]. Macrophages are polarized to either the proinflammatory M1 phenotype or the anti-inflammatory M2 phenotype, which correlates with their contribution to disease progression. It is necessary to find out whether different macrophage phenotypes are the cause of the disease or they simply reflect its progression[132].
At the stage with progressive necrotic lesions, macrophage apoptosis can be partially induced by FC or FA[125]. Macrophages exposed to ER stress are more receptive to apoptosis caused by oxidized phospholipids or LPs[122]. Apoptosis of macrophages does not cause plaque necrosis. This occurs when phagocytes are unable to remove macrophages (efferocytosis) that have undergone apoptosis[133]. Efferocytosis is carried out through phagocytic receptors, ligands of apoptotic cells and bridging molecules. The defect of efferocytosis can occur due to a large number of apoptotic macrophages and tissue necrosis[134]. Other causes of defective efferocytosis may be the death of efferocytes caused by oxidative stress[135] and protease-mediated cleavage of the efferocytosis receptor MerTK (MER tyrosine kinase)[136].
The stage of resolution of atherosclerotic lesions may be caused by an aggressive decrease in lipid levels in mice with hyperlipidemia and a decrease in blood glucose levels in mice with diabetes. Regression in these models is indicated by a decrease in the number of macrophages in the atherosclerotic plaque and a change in gene expression in CD68-positive cells[137]. A decrease in the number of macrophages can occur due to an increase in the effectiveness of efferocytosis and autophagy[125]. With a decrease in the production of collagen of fibrous thickening by fibromyoblast-like SMCs, plaques prone to rupture will appear in the intima[138]. Macrophages can reduce the production of collagen in the SMCs intima, while the cells do not die. In plaques where the balance of apoptotic cells is disturbed, the secretion of TGFß is low. This may be the reason for its loss by neighboring cells[139].
The influence of M1 and M2 macrophages on atherogenesis can be understood by studying the transcription programs, thanks to which their division into phenotypes occurs[140]. Deletion of transcription factor NR4A1 (nuclear receptor subfamily 4 group A member 1) provides the formation of M1 macrophages and rapid progression of atherosclerosis in Apoe-/- and Ldlr-/-mice[141]. The progression of atherosclerosis in the model with Apoe-/-mice is influenced by the directed deletion of transcription factor SP4. This factor stops the differentiation of monocytes towards the M1 phenotype and accelerates the formation of foam cells[142]. Injection of M2-polarizing cytokine IL-13 into Ldlr-/- mice inhibits the progression of atherosclerosis[143].
It has been suggested that several other populations of macrophages can be found in plaques. These macrophage phenotypes are called M(Hb) and Them[144]. They are characterized by resistance to lipid loading. Differentiation towards these phenotypes occurs under the influence of hemoglobin-haptoglobin complexes and heme in vitro. Mox-type macrophages are characterized by high production of hemoxygenase-1[145]. The chemokine CXCL4 leads to the formation of an M4 population[146]. It has been suggested that IL-17A can also influence the polarization of macrophages; as a result of its influence, a population, unlike M1, M2 and M4, is formed[147].
A large number of macrophages in unstable plaques makes them an important subject of research that can help in the diagnosis and therapy of diseases[148]. Macrophages in plaques can be detected by SPIOs (supermagnetic iron oxide particles). Nanoparticles stuck together inside phagolysosomes give a strong MRI (magnetic resonance imaging) signal. This method of macrophage visualization can be used both in experimental models and in patients[149]. This method can help to see how quickly the development of atherosclerotic plaque occurs. The more the particles absorb newly migrating cells, the bigger they become and the more acute the inflammation[150].
In addition, macrophages have the ability to absorb nuclear agents, such as 64Cu-labeled nanoparticles. After that, they can be found using positron emission tomography (PET)[151]. The approximate number of macrophages can be determined using radioactive fluorodeoxyglucose (18F-FDG), after obtaining the result using PET. Cells absorb it instead of the usual glucose[152]. Using the mitochondrial protein (the ligand of the peripheral benzodiazepine receptor), the activity of macrophages can be studied, since a large amount of it is expressed in them[153]. Active work is underway to develop a method by which it will be possible to study individual subpopulations of macrophages, for example, M2[154]. If this method can be applied clinically to better monitor plaque vulnerability, it would be a promising area for future research[155].
Statins can reduce high cholesterol and suppress inflammatory macrophages[156]. In an experiment with statins, their intake increased the number of macrophages in the lesion, and there was also a decrease in other markers of inflammation[157]. FTY720 (biologically active sphingolipid) increases the number of M2 macrophages in plaques and slows down the development of lesions in mice, which indicates an important potential for therapy[158,159].
CONCLUSION
It is necessary to better understand how the metabolism of monocytes and macrophages changes at the stage of initiation of atherosclerotic lesions, and to find out at which point pathological changes occur in cells that contribute to the progression of the disease. These mechanisms require a detailed study of the plasticity of the macrophage phenotype at various stages of atherosclerosis in order to resolve inflammatory reactions and restore protective immune functions. It is necessary to investigate the mechanisms that underlie the relationship between inflammatory processes in rheumatic diseases and the risk of developing CVD in order to create new approaches to treatment and prevention. Studies can be conducted to evaluate the relationship between MPO activity, HDL function, and cholesterol efflux from cells with indicators of subclinical atherosclerosis in rheumatological diseases. It is important to assess the effect of autoimmune diseases on HDL levels and, as a consequence, on the path of cholesterol efflux. Further research on the role of lipid metabolism in the pathogenesis of autoimmune diseases may open up alternative strategies for improving immune cell function. Thus, this knowledge may be of fundamental importance for identifying new approaches in diagnosis and treatment, by studying the mechanisms influencing the lipid content in macrophages, their level and inflammatory phenotype.
DECLARATIONS
Authors’ contributionsText preparation: Bogatyreva AI
Editing: Tolstik TV, Khotina VA
Consultation, English improvement: Markin AM
Manuscript editing: Grechko AV, Oishi Y
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe work was supported by the Ministry of Science and Higher Education of the Russian Federation, Project AAAA-A20-120122190024-0 “Regulation of mitochondrial functional activity by molecular genetic methods, preparation of methodological content for Gene therapy of socially significant diseases - atherosclerosis and essential hypertension”.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Simon TA, Kawabata H, Ray N, Baheti A, Suissa S, Esdaile JM. Prevalence of co-existing autoimmune disease in rheumatoid arthritis: a cross-sectional study. Adv Ther 2017;34:2481-90.
2. Chen W, Wang Q, Zhou B, et al. Lipid metabolism profiles in rheumatic diseases. Front Pharmacol 2021;12:443.
3. Rhoads JP, Major AS, Rathmell JC. Fine tuning of immunometabolism for the treatment of rheumatic diseases. Nat Rev Rheumatol 2017;13:313-20.
4. Sobenin IA, Sazonova MA, Postnov AY, Bobryshev YV, Orekhov AN. Mitochondrial mutations are associated with atherosclerotic lesions in the human aorta. Clin Dev Immunol 2012;2012:832464.
5. Cleeman JI. Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA 2001;285:2486-97.
6. Tselios K, Koumaras C, Gladman DD, Urowitz MB. Dyslipidemia in systemic lupus erythematosus: just another comorbidity? Semin Arthritis Rheum 2016;45:604-10.
7. Hedar AM, Stradner MH, Roessler A, Goswami N. Autoimmune rheumatic diseases and vascular function: the concept of autoimmune atherosclerosis. J Clin Med 2021;10:4427.
9. England BR, Thiele GM, Anderson DR, Mikuls TR. Increased cardiovascular risk in rheumatoid arthritis: mechanisms and implications. BMJ 2018;361:k1036.
10. Sobenin IA, Sazonova MA, Postnov AY, Salonen JT, Bobryshev YV, Orekhov AN. Association of mitochondrial genetic variation with carotid atherosclerosis. PLoS One 2013;8:e68070.
11. Schwartz DM, Burma AM, Kitakule MM, Luo Y, Mehta NN. T Cells in autoimmunity-associated cardiovascular diseases. Front Immunol 2020;11:588776.
12. Sobenin IA, Mitrofanov KY, Zhelankin AV, et al. Quantitative assessment of heteroplasmy of mitochondrial genome: perspectives in diagnostics and methodological pitfalls. Biomed Res Int 2014;2014:292017.
14. Pan L, Lu MP, Wang JH, Xu M, Yang SR. Immunological pathogenesis and treatment of systemic lupus erythematosus. World J Pediatr 2020;16:19-30.
15. Kostopoulou M, Nikolopoulos D, Parodis I, Bertsias G. Cardiovascular disease in systemic lupus erythematosus: recent data on epidemiology, risk factors and prevention. Curr Vasc Pharmacol 2020;18:549-65.
16. Samuelsson I, Parodis I, Gunnarsson I, et al. Myocardial infarctions, subtypes and coronary atherosclerosis in SLE: a case-control study. Lupus Sci Med 2021;8:e000515.
17. Ryu H, Kim J, Kim D, Lee JE, Chung Y. Cellular and molecular links between autoimmunity and lipid metabolism. Mol Cells 2019;42:747-54.
18. Kobiyama K, Ley K. Atherosclerosis: a chronic inflammatory disease with an autoimmune component. Circ Res 2018;123:1118-20.
19. Sobenin IA. Atherogenesis, atherosclerosis and related diseases: unresolved issues. Vessel Plus 2020;4:18.
20. Summerhill VI, Grechko AV, Yet SF, Sobenin IA, Orekhov AN. The atherogenic role of circulating modified lipids in atherosclerosis. Int J Mol Sci 2019;20:3561.
21. Nordestgaard BG. Triglyceride-rich lipoproteins and atherosclerotic cardiovascular disease: new insights from epidemiology, genetics, and biology. Circ Res 2016;118:547-63.
22. Tektonidou MG, Kravvariti E, Konstantonis G, Tentolouris N, Sfikakis PP, Protogerou A. Subclinical atherosclerosis in systemic lupus erythematosus: comparable risk with diabetes mellitus and rheumatoid arthritis. Autoimmun Rev 2017;16:308-12.
23. Chistiakov DA, Sobenin IA, Orekhov AN. Strategies to deliver microRNAs as potential therapeutics in the treatment of cardiovascular pathology. Drug Deliv 2012;19:392-405.
24. Ouimet M, Barrett TJ, Fisher EA. HDL and reverse cholesterol transport. Circ Res 2019;124:1505-18.
25. Goldberg IJ, Reue K, Abumrad NA, et al. Deciphering the role of lipid droplets in cardiovascular disease: a report from the 2017 national heart, lung, and blood institute workshop. Circulation 2018;138:305-15.
26. Brown MS, Goldstein JL, Krieger M, Ho YK, Anderson RG. Reversible accumulation of cholesteryl esters in macrophages incubated with acetylated lipoproteins. J Cell Biol 1979;82:597-613.
27. Guo S, Li L, Yin H. Cholesterol homeostasis and liver X receptor (LXR) in atherosclerosis. Cardiovasc Hematol Disord Drug Targets 2018;18:27-33.
28. Yu XH, Tang CK. ABCA1, ABCG1, and cholesterol homeostasis. Adv Experimental Med Biol 2022;1377:95-107.
29. Allahverdian S, Chaabane C, Boukais K, Francis GA, Bochaton-Piallat ML. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res 2018;114:540-50.
30. Chen W, Li L, Wang J, et al. The ABCA1-efferocytosis axis: a new strategy to protect against atherosclerosis. Clin Chim Acta 2021;518:1-8.
31. Skarda L, Kowal J, Locher KP. Structure of the human cholesterol transporter ABCG1. J Mol Biol 2021;433:167218.
32. Wróblewska M. The origin and metabolism of a nascent pre-β high density lipoprotein involved in cellular cholesterol efflux. Acta Biochim Pol 2011;58:275-85.
33. Shen WJ, Azhar S, Kraemer FB. SR-B1: a unique multifunctional receptor for cholesterol influx and efflux. Annu Rev Physiol 2018;80:95-116.
34. Madison BB. Srebp2: a master regulator of sterol and fatty acid synthesis. J Lipid Res 2016;57:333-5.
35. Wang B, Tontonoz P. Liver X receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol 2018;14:452-63.
36. Hua X, Yokoyama C, Wu J, et al. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci USA 1993;90:11603-7.
37. Kong M, Zhu Y, Shao J, Fan Z, Xu Y. The chromatin remodeling protein BRG1 regulates SREBP maturation by activating SCAP transcription in hepatocytes. Front Cell Dev Biol 2021;9:622866.
38. Duan Y, Gong K, Xu S, Zhang F, Meng X, Han J. Regulation of cholesterol homeostasis in health and diseases: from mechanisms to targeted therapeutics. Signal Transduct Target Ther 2022;7:265.
39. Guo C, Chi Z, Jiang D, et al. Cholesterol homeostatic regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity 2018;49:842-856.e7.
40. Brown MS, Radhakrishnan A, Goldstein JL. Retrospective on cholesterol homeostasis: the central role of scap. Annu Rev Biochem 2018;87:783-807.
41. He C, Hu X, Weston TA, et al. Macrophages release plasma membrane-derived particles rich in accessible cholesterol. Proc Natl Acad Sci USA 2018;115:E8499-508.
42. Hafiane A, Genest J. ATP binding cassette A1 (ABCA1) mediates microparticle formation during high-density lipoprotein (HDL) biogenesis. Atherosclerosis 2017;257:90-9.
43. Ong DS, Anzinger JJ, Leyva FJ, Rubin N, Addadi L, Kruth HS. Extracellular cholesterol-rich microdomains generated by human macrophages and their potential function in reverse cholesterol transport. J Lipid Res 2010;51:2303-13.
44. Jin X, Dimitriadis EK, Liu Y, et al. Macrophages shed excess cholesterol in unique extracellular structures containing cholesterol microdomains. Arterioscler Thromb Vasc Biol 2018;38:1504-18.
45. He C, Hu X, Weston TA, et al. Macrophages release plasma membrane-derived particles rich in accessible cholesterol. Proc Natl Acad Sci USA 2018;115:E8499-508.
46. Hu X, Weston TA, He C, et al. Release of cholesterol-rich particles from the macrophage plasma membrane during movement of filopodia and lamellipodia. Elife 2019;8:e50231.
47. He C, Jiang H, Song W, et al. Cultured macrophages transfer surplus cholesterol into adjacent cells in the absence of serum or high-density lipoproteins. Proc Natl Acad Sci USA 2020;117:10476-83.
48. Dupont M, Souriant S, Lugo-Villarino G, Maridonneau-Parini I, Vérollet C. Tunneling nanotubes: intimate communication between myeloid cells. Front Immunol 2018;9:43.
49. Kentala H, Weber-Boyvat M, Olkkonen VM. OSBP-related protein family: mediators of lipid transport and signaling at membrane contact sites. Int Rev Cell Mol Biol 2016;321:299-340.
50. Ouimet M, Hennessy EJ, van Solingen C, et al. miRNA targeting of oxysterol-binding protein-like 6 regulates cholesterol trafficking and efflux. Arterioscler Thromb Vasc Biol 2016;36:942-51.
51. Schulman IG. Liver X receptors link lipid metabolism and inflammation. FEBS Lett 2017;591:2978-91.
52. Endo-Umeda K, Kim E, Thomas DG, et al. Myeloid LXR (liver X receptor) deficiency induces inflammatory gene expression in foamy macrophages and accelerates atherosclerosis. Arterioscler Thromb Vasc Biol 2022;42:719-31.
53. Teupser D, Kretzschmar D, Tennert C, et al. Effect of macrophage overexpression of murine liver X receptor-alpha (LXR-alpha) on atherosclerosis in LDL-receptor deficient mice. Arterioscler Thromb Vasc Biol 2008;28:2009-15.
54. Savla SR, Prabhavalkar KS, Bhatt LK. Liver X receptor: a potential target in the treatment of atherosclerosis. Expert Opin Ther Targets 2022;26:645-58.
55. Saliba-Gustafsson P, Pedrelli M, Gertow K, et al. Subclinical atherosclerosis and its progression are modulated by PLIN2 through a feed-forward loop between LXR and autophagy. J Intern Med 2019;286:660-75.
56. Groh L, Keating ST, Joosten LAB, Netea MG, Riksen NP. Monocyte and macrophage immunometabolism in atherosclerosis. Semin Immunopathol 2018;40:203-14.
57. Bes C, Gürel S, Buğdaycı G, Dikbaş O, Soy M. Atherosclerosis assessment and rheumatoid arthritis. Z Rheumatol 2018;77:330-4.
58. Kraakman MJ, Dragoljevic D, Kammoun HL, Murphy AJ. Is the risk of cardiovascular disease altered with anti-inflammatory therapies? Clin Transl Immunology 2016;5:e84.
59. Hirose S, Lin Q, Ohtsuji M, Nishimura H, Verbeek JS. Monocyte subsets involved in the development of systemic lupus erythematosus and rheumatoid arthritis. Int Immunol 2019;31:687-96.
60. Cook AD, Louis C, Robinson MJ, Saleh R, Sleeman MA, Hamilton JA. Granulocyte macrophage colony-stimulating factor receptor α expression and its targeting in antigen-induced arthritis and inflammation. Arthritis Res Ther 2016;18:287.
61. Murphy AJ, Akhtari M, Tolani S, et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest 2011;121:4138-49.
62. Quevedo-Abeledo JC, Sánchez-Pérez H, Tejera-Segura B, et al. Differences in capacity of high-density lipoprotein cholesterol efflux between patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Care Res 2021;73:1590-6.
63. Ormseth MJ, Yancey PG, Yamamoto S, et al. Net cholesterol efflux capacity of HDL enriched serum and coronary atherosclerosis in rheumatoid arthritis. IJC Metab Endocr 2016;13:6-11.
64. Xie B, He J, Liu Y, Liu T, Liu C. A meta-analysis of HDL cholesterol efflux capacity and concentration in patients with rheumatoid arthritis. Lipids Health Dis 2021;20:18.
65. Nowak B, Madej M, Łuczak A, Małecki R, Wiland P. Disease activity, oxidized-LDL fraction and anti-oxidized LDL antibodies influence cardiovascular risk in rheumatoid arthritis. Adv Clin Exp Med 2016;25:43-50.
66. Westerterp M, Tsuchiya K, Tattersall IW, et al. Deficiency of ATP-binding cassette transporters A1 and G1 in endothelial cells accelerates atherosclerosis in mice. Arterioscler Thromb Vasc Biol 2016;36:1328-37.
67. Hannawi S, Hannawi H, Al Salmi I. Cardiovascular disease and subclinical atherosclerosis in rheumatoid arthritis. Hypertens Res 2020;43:982-4.
68. Dragoljevic D, Kraakman MJ, Nagareddy PR, et al. Defective cholesterol metabolism in haematopoietic stem cells promotes monocyte-driven atherosclerosis in rheumatoid arthritis. Eur Heart J 2018;39:2158-67.
69. Charles-Schoeman C, Lee YY, Grijalva V, et al. Cholesterol efflux by high density lipoproteins is impaired in patients with active rheumatoid arthritis. Ann Rheum Dis 2012;71:1157-62.
70. Aratani Y. Myeloperoxidase: its role for host defense, inflammation, and neutrophil function. Arch Biochem Biophys 2018;640:47-52.
71. Frangie C, Daher J. Role of myeloperoxidase in inflammation and atherosclerosis (Review). Biomed Rep 2022;16:53.
72. Ndrepepa G. Myeloperoxidase - a bridge linking inflammation and oxidative stress with cardiovascular disease. Clin Chim Acta 2019;493:36-51.
73. Zheng L, Nukuna B, Brennan ML, et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest 2004;114:529-41.
74. Steiner G, Urowitz MB. Lipid profiles in patients with rheumatoid arthritis: mechanisms and the impact of treatment. Semin Arthritis Rheum 2009;38:372-81.
75. MacLeod C, Hadoke PWF, Nixon M. Glucocorticoids: fuelling the Fire of atherosclerosis or therapeutic extinguishers? Int J Mol Sci 2021;22:7622.
76. Liu T, Shi N, Zhang S, et al. Systemic lupus erythematosus aggravates atherosclerosis by promoting IgG deposition and inflammatory cell imbalance. Lupus 2020;29:273-82.
77. Lerang K, Gilboe IM, Steinar Thelle D, Gran JT. Mortality and years of potential life loss in systemic lupus erythematosus: a population-based cohort study. Lupus 2014;23:1546-52.
78. Chiesa ST, Charakida M. High-density lipoprotein function and dysfunction in health and disease. Cardiovasc Drugs Ther 2019;33:207-19.
79. Wang Y, Yu H, He J. Role of dyslipidemia in accelerating inflammation, autoimmunity, and atherosclerosis in systemic lupus erythematosus and other autoimmune diseases. Discov Med 2020;30:49-56.
80. Kim SY, Yu M, Morin EE, Kang J, Kaplan MJ, Schwendeman A. High-density lipoprotein in lupus: disease biomarkers and potential therapeutic strategy. Arthritis Rheumatol 2020;72:20-30.
81. Aguilar-Ballester M, Herrero-Cervera A, Vinué Á, Martínez-Hervás S, González-Navarro H. Impact of cholesterol metabolism in immune cell function and atherosclerosis. Nutrients 2020;12:2021.
82. Szabó MZ, Szodoray P, Kiss E. Dyslipidemia in systemic lupus erythematosus. Immunol Res 2017;65:543-50.
83. Shridas P, Tannock LR. Role of serum amyloid A in atherosclerosis. Curr Opin Lipidol 2019;30:320-5.
84. Jin Z, Zhou L, Tian R, Lu N. Myeloperoxidase targets apolipoprotein A-I for site-specific tyrosine chlorination in atherosclerotic lesions and generates dysfunctional high-density lipoprotein. Chem Res Toxicol 2021;34:1672-80.
85. Witkowski A, Chan GKL, Boatz JC, et al. Methionine oxidized apolipoprotein A-I at the crossroads of HDL biogenesis and amyloid formation. FASEB J 2018;32:3149-65.
86. Xepapadaki E, Zvintzou E, Kalogeropoulou C, Filou S, Kypreos KE. Τhe antioxidant function of HDL in atherosclerosis. Angiology 2020;71:112-21.
87. De Nardo D, Labzin LI, Kono H, et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat Immunol 2014;15:152-60.
88. Yin K, Chen WJ, Zhou ZG, et al. Apolipoprotein A-I inhibits CD40 proinflammatory signaling via ATP-binding cassette transporter A1-mediated modulation of lipid raft in macrophages. J Atheroscler Thromb 2012;19:823-36.
89. Parra S, Vives G, Ferré R, et al. Complement system and small HDL particles are associated with subclinical atherosclerosis in SLE patients. Atherosclerosis 2012;225:224-30.
90. Skaggs BJ, Hahn BH, Sahakian L, Grossman J, McMahon M. Dysfunctional, pro-inflammatory HDL directly upregulates monocyte PDGFRβ, chemotaxis and TNFα production. Clin Immunol 2010;137:147-56.
91. Markin A, Markina Y, Sukhorukov V, et al. The role of physical activity in the development of atherosclerotic lesions of the vascular wall. Clin Exp Morphol 2019;8:25-31.
92. Smith CK, Seto NL, Vivekanandan-Giri A, et al. Lupus high-density lipoprotein induces proinflammatory responses in macrophages by binding lectin-like oxidised low-density lipoprotein receptor 1 and failing to promote activating transcription factor 3 activity. Ann Rheum Dis 2017;76:602-11.
94. Lewis MJ, Malik TH, Fossati-Jimack L, et al. Distinct roles for complement in glomerulonephritis and atherosclerosis revealed in mice with a combination of lupus and hyperlipidemia. Arthritis Rheum 2012;64:2707-18.
95. Robinson G, Pineda-Torra I, Ciurtin C, Jury EC. Lipid metabolism in autoimmune rheumatic disease: implications for modern and conventional therapies. J Clin Invest 2022;132:e148552.
96. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol 2015;15:511-23.
97. Howie D, Ten Bokum A, Necula AS, Cobbold SP, Waldmann H. The role of lipid metabolism in T lymphocyte differentiation and survival. Front Immunol 2017;8:1949.
98. Zhornitsky S, McKay KA, Metz LM, Teunissen CE, Rangachari M. Cholesterol and markers of cholesterol turnover in multiple sclerosis: relationship with disease outcomes. Mult Scler Relat Disord 2016;5:53-65.
99. Sun W, Li P, Cai J, et al. Lipid metabolism: immune regulation and therapeutic prospectives in systemic lupus erythematosus. Front Immunol 2022;13:1154.
100. Shih CM, Chen CC, Chu CK, Wang KH, Huang CY, Lee AW. The roles of lipoprotein in psoriasis. Int J Mol Sci 2020;21:859.
101. Nowowiejska J, Baran A, Flisiak I. Aberrations in lipid expression and metabolism in psoriasis. Int J Mol Sci 2021;22:6561.
102. Masson W, Lobo M, Molinero G. Psoriasis and cardiovascular risk: a comprehensive review. Adv Ther 2020;37:2017-33.
103. Ramezani M, Zavattaro E, Sadeghi M. Evaluation of serum lipid, lipoprotein, and apolipoprotein levels in psoriatic patients: a systematic review and meta-analysis of case-control studies. Postepy Dermatol Alergol 2019;36:692-702.
104. Miller IM, Skaaby T, Ellervik C, Jemec GB. Quantifying cardiovascular disease risk factors in patients with psoriasis: a meta-analysis. Br J Dermatol 2013;169:1180-7.
105. Holzer M, Wolf P, Curcic S, et al. Psoriasis alters HDL composition and cholesterol efflux capacity. J Lipid Res 2012;53:1618-24.
106. Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol 1995;15:551-61.
107. Yang H, Zhang N, Okoro EU, Guo Z. Transport of apolipoprotein B-containing lipoproteins through endothelial cells is associated with apolipoprotein E-carrying HDL-like particle formation. Int J Mol Sci 2018;19:3593.
108. Tiwari S, Siddiqi SA. Intracellular trafficking and secretion of very low density lipoproteins. Arterioscler Thromb Vasc Biol 2012;32:1079-86.
109. Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med 2008;18:228-32.
110. Frambach SJCM, de Haas R, Smeitink JAM, Rongen GA, Russel FGM, Schirris TJJ. Brothers in arms: ABCA1- and ABCG1-Mediated cholesterol efflux as promising targets in cardiovascular disease treatment. Pharmacol Rev 2020;72:152-90.
112. Soldatov VO, Malorodova TN, Pokrovskaya TG, et al. Ultrasonic dopplerography for the evaluation of endothelial function in the conduct of pharmacological vascular samples in an experiment Production and Hosted by. Int J Res Pharm Sci 2018;9:735-40.
113. Filippi MD. Mechanism of diapedesis: importance of the transcellular route. Adv Immunol 2016;129:25-53.
114. Soldatov VO, Malorodova TN, Balamutova TI, Ksenofontov AO, Dovgan AP, Urozhevskaya ZS. Endothelial dysfunction: comparative evaluation of ultrasound dopplerography, laser dopplerflowmetry and direct monitoring of arterial pressure for conducting pharmacological tests in rats. Res Results Pharmacol 2018;4:73-80.
115. Liberale L, Dallegri F, Montecucco F, Carbone F. Pathophysiological relevance of macrophage subsets in atherogenesis. Thromb Haemost 2017;117:7-18.
116. Chistiakov DA, Orekhov AN, Sobenin IA, Bobryshev YV. Plasmacytoid dendritic cells: development, functions, and role in atherosclerotic inflammation. Front Physiol 2014;5:279.
117. Orekhov AN, Myasoedova VA. Low density lipoprotein-induced lipid accumulation is a key phenomenon of atherogenesis at the arterial cell level. Vessel Plus 2019;3:3.
118. Chistiakov DA, Sobenin IA, Orekhov AN, Bobryshev YV. Myeloid dendritic cells: development, functions, and role in atherosclerotic inflammation. Immunobiology 2015;220:833-44.
119. Yan J, Horng T. Lipid metabolism in regulation of macrophage functions. Trends Cell Biol 2020;30:979-89.
120. Paukner K, Králová Lesná I, Poledne R. Cholesterol in the cell membrane-an emerging player in atherogenesis. Int J Mol Sci 2022;23:533.
121. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997;89:331-40.
122. Boucher P, Matz RL, Terrand J. atherosclerosis: gone with the Wnt? Atherosclerosis 2020;301:15-22.
123. Groenen AG, Halmos B, Tall AR, Westerterp M. Cholesterol efflux pathways, inflammation, and atherosclerosis. Crit Rev Biochem Mol Biol 2021;56:426-39.
124. Ikonen E, Zhou X. Cholesterol transport between cellular membranes: a balancing act between interconnected lipid fluxes. Dev Cell 2021;56:1430-6.
125. Kim KW, Ivanov S, Williams JW. Monocyte recruitment, specification, and function in atherosclerosis. Cells 2020;10:15.
126. Moroni F, Ammirati E, Norata GD, Magnoni M, Camici PG. The role of monocytes and macrophages in human atherosclerosis, plaque neoangiogenesis, and atherothrombosis. Mediators Inflamm 2019;2019:7434376.
127. Baidžajevas K, Hadadi É, Lee B, et al. Macrophage polarisation associated with atherosclerosis differentially affects their capacity to handle lipids. Atherosclerosis 2020;305:10-8.
128. Qiao JH, Tripathi J, Mishra NK, et al. Role of macrophage colony-stimulating factor in atherosclerosis: studies of osteopetrotic mice. Am J Pathol 1997;150:1687.
129. Manjarrez-Reyna AN, Martínez-Reyes CP, Aguayo-Guerrero JA, et al. Native low-density lipoproteins act in synergy with lipopolysaccharide to alter the balance of human monocyte subsets and their ability to produce IL-1 beta, CCR2, and CX3CR1 in vitro and in vivo: implications in atherogenesis. Biomolecules 2021;11:1169.
130. Chistiakov DA, Revin VV, Sobenin IA, Orekhov AN, Bobryshev YV. Vascular endothelium: functioning in norm, changes in atherosclerosis and current dietary approaches to improve endothelial function. Mini Rev Med Chem 2015;15:338-50.
131. Sobenin IA, Sazonova MA, Postnov AY, Bobryshev YV, Orekhov AN. Changes of mitochondria in atherosclerosis: possible determinant in the pathogenesis of the disease. Atherosclerosis 2013;227:283-8.
132. Yang S, Yuan HQ, Hao YM, et al. Macrophage polarization in atherosclerosis. Clin Chim Acta 2020;501:142-6.
133. Dhawan UK, Singhal A, Subramanian M. Dead cell and debris clearance in the atherosclerotic plaque: Mechanisms and therapeutic opportunities to promote inflammation resolution. Pharmacol Res 2021;170:105699.
134. Shklover J, Levy-Adam F, Kurant E. Apoptotic cell clearance in development. Curr Top Dev Biol 2015;114:297-334.
135. Zhao Y, Zhang L, Liu L, et al. Specific loss of ABCA1 (ATP-binding cassette transporter A1) suppresses TCR (T-cell receptor) signaling and provides protection against atherosclerosis. Arterioscler Thromb Vasc Biol 2022;42:e311-26.
136. Wanke F, Gutbier S, Rümmelin A, et al. Ligand-dependent kinase activity of MERTK drives efferocytosis in human iPSC-derived macrophages. Cell Death Dis 2021;12:538.
137. Ruotsalainen AK, Lappalainen JP, Heiskanen E, et al. Nuclear factor E2-related factor 2 deficiency impairs atherosclerotic lesion development but promotes features of plaque instability in hypercholesterolaemic mice. Cardiovasc Res 2019;115:243-54.
138. Boyle JJ, Weissberg PL, Bennett MR. Tumor necrosis factor-alpha promotes macrophage-induced vascular smooth muscle cell apoptosis by direct and autocrine mechanisms. Arterioscler Thromb Vasc Biol 2003;23:1553-8.
139. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 1998;101:890-8.
140. Orekhov AN, Orekhova VA, Nikiforov NG, et al. Monocyte differentiation and macrophage polarization. Vessel Plus 2019;3:10.
141. Hanna RN, Carlin LM, Hubbeling HG, et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat Immunol 2011;12:778-85.
142. Tang RZ, Zhu JJ, Yang FF, et al. DNA methyltransferase 1 and Krüppel-like factor 4 axis regulates macrophage inflammation and atherosclerosis. J Mol Cell Cardiol 2019;128:11-24.
143. Cardilo-Reis L, Gruber S, Schreier SM, et al. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol Med 2012;4:1072-86.
144. Mushenkova NV, Nikiforov NG, Melnichenko AA, et al. Functional phenotypes of intraplaque macrophages and their distinct roles in atherosclerosis development and atheroinflammation. Biomedicines 2022;10:452.
145. Kadl A, Meher AK, Sharma PR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res 2010;107:737-46.
146. Jinnouchi H, Guo L, Sakamoto A, et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell Mol Life Sci 2020;77:1919-32.
147. Nordlohne J, von Vietinghoff S. Interleukin 17A in atherosclerosis - regulation and pathophysiologic effector function. Cytokine 2019;122:154089.
148. Yao SY, Shen ML, Li SJ, et al. Application of a mechanically responsive, inflammatory macrophage-targeted dual-sensitive hydrogel drug carrier for atherosclerosis. Colloids Surf B Biointerfaces 2020;186:110718.
149. Chen X, Li X, Chen Q. Experimental study of ultrafine superparamagnetic iron oxide-enhanced MRI in an atherosclerotic plaque model. J Nanosci Nanotechnol 2020;20:7444-50.
150. Li Y, Pan Y, Wu X, et al. Dual-modality imaging of atherosclerotic plaques using ultrasmall superparamagnetic iron oxide labeled with rhodamine. Nanomedicine 2019;14:1935-44.
151. Nahrendorf M, Hoyer FF, Meerwaldt AE, et al. Imaging Cardiovascular and lung macrophages with the positron emission tomography sensor 64Cu-macrin in mice, rabbits, and pigs. Circ Cardiovasc Imaging 2020;13:e010586.
152. Konishi T, Norikane T, Yamamoto Y, et al. The potential relationship between 18F-FDG uptake and wall shear stress in a patient with carotid artery disease. J Nucl Cardiol 2021;28:367-70.
153. Fujimura Y, Hwang PM, Trout Iii H, et al. Increased peripheral benzodiazepine receptors in arterial plaque of patients with atherosclerosis: an autoradiographic study with [(3)H]PK 11195. Atherosclerosis 2008;201:108-11.
154. Han S, Wang W, Wang S, et al. Tumor microenvironment remodeling and tumor therapy based on M2-like tumor associated macrophage-targeting nano-complexes. Theranostics 2021;11:2892-916.
155. Taghizadeh E, Taheri F, Renani PG, Reiner Ž, Navashenaq JG, Sahebkar A. Macrophage: a key therapeutic target in atherosclerosis? Curr Pharm Des 2019;25:3165-74.
156. Hetherington I, Totary-Jain H. Anti-atherosclerotic therapies: milestones, challenges, and emerging innovations. Mol Ther 2022;30:3106-17.
157. Kang MK, Kim CJ, Choo EH, et al. Anti-inflammatory effect of statin is continuously working throughout use: a prospective three time point 18F-FDG PET/CT imaging study. Int J Cardiovasc Imaging 2019;35:1745-53.
158. Nofer JR, Bot M, Brodde M, et al. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2007;115:501-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Bogatyreva AI, Tolstik TV, Khotina VA, Grechko AV, Oishi Y, Markin AM. Features of cholesterol metabolism in macrophages in immunoinflammatory diseases. Vessel Plus 2023;7:4. http://dx.doi.org/10.20517/2574-1209.2022.24
AMA Style
Bogatyreva AI, Tolstik TV, Khotina VA, Grechko AV, Oishi Y, Markin AM. Features of cholesterol metabolism in macrophages in immunoinflammatory diseases. Vessel Plus. 2023; 7: 4. http://dx.doi.org/10.20517/2574-1209.2022.24
Chicago/Turabian Style
Bogatyreva, Anastasia I., Taisiya V. Tolstik, Victoria A. Khotina, Andrey V. Grechko, Yumiko Oishi, Alexander M. Markin. 2023. "Features of cholesterol metabolism in macrophages in immunoinflammatory diseases" Vessel Plus. 7: 4. http://dx.doi.org/10.20517/2574-1209.2022.24
ACS Style
Bogatyreva, AI.; Tolstik TV.; Khotina VA.; Grechko AV.; Oishi Y.; Markin AM. Features of cholesterol metabolism in macrophages in immunoinflammatory diseases. Vessel Plus. 2023, 7, 4. http://dx.doi.org/10.20517/2574-1209.2022.24
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 16 clicks
Cite This Article 16 clicks

 Like This Article 29
likes
Like This Article 29
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.