
Challenges of mitochondrial DNA editing in mammalian cells: focus on treatment of cardiovascular disease
 0
0 , ...
, ... Abstract
Atherosclerosis is the major cause of occurrence and development of cardiovascular disease. Mutations in mitochondrial DNA (mtDNA) can lead to the development of several pathologies. Over the last few years, there has been increasing evidence that mitochondrial dysfunction caused by mtDNA mutations is associated with atherogenesis and other diseases of the cardiovascular system. Several therapeutic approaches have been developed for the improvement of mitochondrial function, and they are mainly associated with the cellular and tissue antioxidant defense system. However, these approaches are not targeted at mtDNA mutations, which trigger the pathogenesis of disease. Gene-editing technologies could be a promising approach for the treatment of cardiovascular disease caused by mtDNA mutations. To date, such technologies have shown considerable success in mitochondrial gene editing in cell and animal models. Gene-editing technologies allow the determination of the role of mitochondrial genome mutations in the development and complication of various chronic diseases. Nevertheless, further investigation and optimization in this field is required for future human trials. This review highlights the progress and existing challenges of modern technologies and approaches to mitochondrial gene editing.
Keywords
INTRODUCTION
A wide range of mutations in the mitochondrial genome causes the development of many human diseases. These diseases include multiple symmetric lipomatosis, mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS), multiple sclerosis (MS), Leber hereditary optic neuropathy (LHON), mitochondrial tubulointerstitial kidney disease, and mitochondrial diabetes and diseases of the cardiovascular system, such as atherosclerosis[1-7]. Atherosclerosis is a multifactorial disease that underlies many pathologies of the cardiovascular system, and at present, remains the leading cause of death worldwide[8]. The development of atherosclerosis is closely associated with the aging process, chronic inflammation, endothelial dysfunction, oxidative stress, and increased plasma lipoprotein concentrations[9-13]. Furthermore, recent studies have demonstrated that atherogenesis may occur owing to damage of mitochondrial DNA (mtDNA) and subsequent mitochondrial dysfunction[14-16].
The methods for mitochondrial genome editing appear to be promising strategies for the treatment of mitochondrial diseases and for atherosclerosis and cardiovascular diseases[17-21]. Most of the currently developed drugs and therapeutic approaches have antioxidant properties and aim to reduce oxidative stress in cells. Nonetheless, the functional role of various mtDNA variants that underlie diseases and the molecular mechanisms that cause mitochondrial dysfunction need to be taken into account to design and develop future therapeutic approaches.
One possible therapeutic approach for the treatment of atherosclerosis might be the application of mitochondrial gene-editing technologies for cells involved in the formation of atherosclerotic lesions. Despite the considerable progress made in mtDNA editing in cellular and animal models of diseases, there are some limitations in targeted delivery of genome editing systems to cells and tissues, and to the mitochondria. To date, these limitations have impeded the application of gene-editing approaches for treating mitochondrial diseases. More research is required to identify risks and solutions to overcome challenges in the implementation of mtDNA gene-editing technologies.
Nevertheless, existing gene-editing technologies can be applied with high efficiency to establish the role of mitochondrial mutations in the development of atherosclerosis[22-24]. The optimization of editing technologies for improving genome editing efficiency in cells involved in the formation of atherosclerotic lesions will contribute to their further use in therapy and treatment of atherosclerosis.
STRUCTURE AND FUNCTION OF MITOCHONDRIAL DNA
Mitochondria are multifunctional organelles that provide eukaryotic cells with the necessary resources for vital functions. Mitochondria are involved in energy metabolism, amino acid, fatty acid, lipid, and nucleotide biosynthesis, and the reactions of the tricarboxylic acid cycle and the ornithine cycle[25,26]. In addition, mitochondria play a major role in programmed cell death by apoptosis and in the regulation of cell survival under physiological and pathological conditions[27]. Furthermore, mitochondria are the source of various pathogen-associated molecular patterns, damage-associated molecular patterns, and reactive oxygen species (ROS) molecules, which trigger innate immune responses that lead to the development of inflammatory processes[14,28-30].
The endosymbiotic theory suggests the origin of mitochondria. According to this theory, the original anaerobic eukaryotic cells engulfed the precursors of mitochondria, which were the ancestors of the Alphaproteobacteria class, during the evolutionary processes[31]. The main evidence of the mitochondrial endosymbiotic origin is as follows. There are two membranes (outer and inner) surrounding the intramitochondrial matrix and intermembrane space. Additionally, the presence of mtDNA, mitochondrial ribosomes (mitoribosomes), cardiolipin in the composition of membranes of bacterial origin, and β-barrel proteins in the outer membrane are further evidence.
Mitochondrial DNA is a circular double-stranded molecule consisting of 16,569 base pairs (bp)[32]. The strands of mtDNA are termed as “heavy” and “light”. Heavy-strand and light-strand mtDNA are transcribed as long polycistronic molecules, where transcription is initiated from the heavy-strand promoter and the light-strand promoter[33]. The mitochondrial genome does not contain introns, although some regions, such as encoding genes, may overlap. Additionally, mtDNA is characterized by multiple copies. Therefore, mtDNA can be present in the body tissue cells in an amount ranging from 100 to 10,000 copies[31]. However, there is one non-coding region in mtDNA called the displacement loop (D-loop), which is a three-stranded structure formed by the inclusion of a short strand of 7S DNA. The D-loop may play a role in nucleotide homeostasis, mtDNA replication, subsequent nucleoid organization, and aging[33]. The
Only 1% of proteins are encoded in the mitochondrial genome, while the rest are encoded in the nuclear genome[26]. Mitochondrial protein synthesis encoded by nuclear DNA is carried out in the cell cytoplasm, and subsequently, its precursors are imported into the mitochondria through translocases, which are localized in the mitochondrial membrane. The mitochondrial protein precursors are then redistributed among intramitochondrial compartments. The synthesis of mitochondrial proteins of the mitochondrial genome occurs owing to the functioning of mitoribosomes[35]. Mitochondrial DNA contains only 37 genes that encode 2 ribosomal RNAs (rRNAs, 12S and 16S), 22 transport RNAs (tRNAs), and 11 messenger RNAs (mRNAs), and 13 proteins. These proteins are mainly represented by subunits of the enzyme complexes I, III, IV, and V of the oxidative phosphorylation system[31,36]. Generally, the human mitochondrial proteome includes approximately 1500 proteins[26]. During the evolutionary process, the genome of the mitochondrial precursor endosymbiont was partially lost or incorporated into the genome of host cells. Therefore, mitochondrial proteins have a dual genetic origin.
Bacterial chromosomes and mtDNA are packaged in a nucleoprotein complex called a nucleoid, where the main structural protein is mitochondrial transcription factor A (TFAM). TFAM is a common protein that also acts as a transcription factor in mitochondria. Nucleoids are approximately
There are several mechanisms responsible for the nuclear-like mtDNA repair mechanism. The most studied mtDNA repair mechanism is base excision repair (BER), which repairs oxidized bases in mtDNA or double-strand breaks (DSBs), resulting from enzymatic processing steps[43,44]. There are two sub-pathways of BER called short-patch BER and long-patch BER. Short-patch BER is characterized by replacing only one abasic site-containing nucleotide, and long-patch BER is capable of replacing 2 to 15 nucleotides in the strand. In the activation of this mechanism, some proteins in mitochondria are also involved in the nuclear mechanism of nucleotide excision repair[45]. However, the presence of mismatch repair and DSB repair mechanisms in mitochondria, such as homologous recombination, non-homologous end-joining, and microhomology-mediated end-joining (MMEJ), is poorly understood. Various studies have suggested the presence of homologous recombination pathways and their predominance over other mechanisms, such as the presence of proteins in the mitochondrial matrix (e.g., Rad52, Rad51, Rad59p, MRE11, and NIBRIN), which carry out similar mechanisms in the nucleus[46-51]. Some studies have also indicated the possible presence of the MMEJ pathway in mitochondria, which is mediated by CtIP, FEN1, MRE11, poly(ADP-ribose) polymerase, and DNA ligase III[52-54]. Currently, there is insufficient direct evidence for the presence of classical non-homologous end-joining and mismatch repair pathways in mitochondria, which are present in nuclear DNA[49,51,52,55,56].
MITOCHONDRIAL DNA MUTATION IS ASSOCIATED WITH ATHEROSCLEROSIS AND CARDIOVASCULAR DISEASE
Mutations in the mitochondrial genome contribute to changes in the structural and functional properties of mitochondria and are the cause of the development of diseases characterized by impaired oxidative phosphorylation. Furthermore, these mutations can underlie the processes involved in the development of cardiovascular diseases, such as atherosclerosis[6,14,57,58]. Mitochondrial genome mutations can be used as a potential biomarker to assess underlying risk factors for the development of pathological conditions [Figure 1].
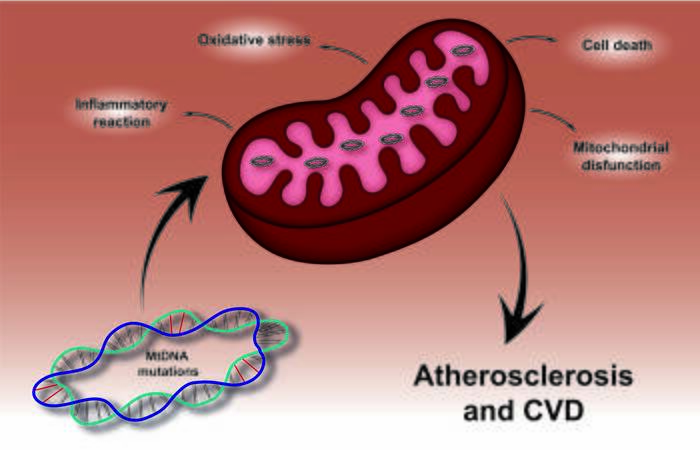
Figure 1. Effect of mtDNA mutations in atherosclerosis and cardiovascular disease. Mutations in the mitochondrial genome lead to mitochondrial dysfunction followed by oxidative stress, accompanied by the development of pro-inflammatory reactions and cell death. Such events play a crucial role in the development and progress of atherosclerosis and cardiovascular disease.
Such mutations in the mitochondrial genome are homoplasmic when they may be present or absent in the genome or heteroplasmic when different proportions of the mutant allele are observed in mtDNA. The level of heteroplasmy is an important factor in determining the amount of mitochondrial dysfunction and the severity of the disease. A threshold of 60%-90% of mutant mtDNA heteroplasmy is required for clinical manifestation[7,14,59]. Qualitative estimations of mutant alleles and a quantitative estimation of the heteroplasmy level in the mitochondrial genome are necessary for studying the association between mitochondrial mutations and human diseases.
Modern methods for detecting and studying mtDNA mutations have shown a correlation between some mutations and various chronic human diseases. In particular, these diseases comprise pathologies of the cardiovascular system, such as coronary stenosis, myocardial infarction, coronary heart disease, cardiomyopathy, atherosclerosis, stroke, and peripheral arterial disease, as well as the development of cancer, and some forms of diabetes and deafness[6,7,14,34,59-61].
Mutations of various tRNAs encoded in the mitochondrial genome are the most commonly studied. Therefore, mutations in mitochondrial tRNAAla (MT-TA) m.5592A>G and tRNAThr (MT-TT) m.15927G>A may be inherited risk factors for CHD[62]. These two mutations can alter tRNA structure and function, leading to mitochondrial dysfunction and a long-term increase in ROS generation in cardiovascular cells. The mutations m.8326A>G, m.8331A>G, m.8324T>A, and m.8344A>G in the tRNALys (MT-TK) gene have also been identified in patients with CHD[63]. In addition, a recently identified m.8231C>A heteroplasmic mutation in the MT-CO2 gene may be involved in the development of CHD[61]. Another report showed two point mutations associated with coronary atherosclerosis, m.5711A>G and m.5725T>G (homo- and heteroplasmic, respectively), in the tRNAAsn (MT-TN) gene, and a homoplasmic m.5568A>G mutation in tRNATrp (MT-TW)[64]. The m.5725T>G mutation in the tRNAAsn gene is a pathogenic mutation in CHD and is highly conserved among species. The m.8357T>C mutation in the gene encoding tRNALys was found in a patient with multiple symmetric lipomatosis[1]. A report showed that 55 heteroplasmic and homoplasmic mutations in the MT-ND1, MT-ND2, tRNAIle (MT-TI), tRNAMet (MT-TM), and tRNAGln (MT-TQ) genes encoded in the mtDNA 3777-4679 region were positively correlated with the manifestation of maternally inherited essential hypertension[65]. These mtDNA mutations include m.3970C>T, m.4048G>A, m.4071C>T, m.4086C>T, m.4164A>G, and m.4248 T>C in MT-ND1 and m.4386T>C and m.4394C>T in MT-TQ, as well as m.4563delG, m.4576delA, m.4611delA, and m.4612delT deletions.
The mitochondrial deletion of 4977 bp (mtDNA4977) has been proposed to be involved in major adverse cardiac events, CHD, and breast cancer. mtDNA4977 can be used as a biomarker of mitochondrial dysfunction and mtDNA oxidative damage. The deletion of mtDNA4977 may affect genes encoding five tRNA genes, four complex I subunits, one complex IV subunit, and two complex V subunits of respiratory chain complexes, and may impair energy production leading to ROS generation[6]. Higher levels of mtDNA4977 deletion in patients with CHD are associated with a reduced mitochondrial DNA copy number and high mortality[66]. Increased mtDNA damage may initially stimulate mitochondrial biogenesis and subsequently increase mitochondrial abundance as an adaptive response. Excessive mtDNA damage can then lead to mtDNA depletion and mitochondrial dysfunction, which play a crucial role in the development and progress of atherosclerosis.
Numerous studies have associated mtDNA mutations with the development of atherosclerosis[7,14,59,67,68]. Their prevalence is affected by various factors, such as sex and regional location[64,69-71]. Whole blood cells and buccal epithelial cells are used as biomarkers of mitochondrial mutations associated with atherosclerosis[72-74]. The detection of threshold heteroplasmy levels of mtDNA mutations, above which patients are at increased risk of atherosclerotic progression, may be a new assessment criterion for the occurrence and development of atherosclerotic lesions in human arteries[73,75].
Positive correlations have been found for heteroplasmy of mtDNA mutations, such as m.15059G>A, m.12315G>A, m.5178C>A, m.3256C>T, m.652delG, m.3336T>C, and m.14459G>A with carotid intima-media thickness and atherosclerosis. Additionally, negative correlations were found for heteroplasmy of m.1555A>G, m.13513G>A, and m.14846G>A mutations[15,69-71,76,77]. However, there are significant differences in the heteroplasmy level between healthy donors and patients with atherosclerosis. These mutations are associated with the mitochondrial genes rRNA 12S (MT-RNR1), 16S RNA (MT-RNR2), tRNALeu (MT-TL1 and MT-TL2), NADH dehydrogenase subunits I, II, V and VI (MT-ND1, MT-ND2, MT-ND5 and MT-ND6, respectively), and cytochrome b (MT-CYB)[7,68,78]. In addition, studies of the correlations between metabolic syndrome and ten mutations associated with atherosclerosis showed that heteroplasmy levels of these mutations were correlated with symptoms of metabolic syndrome, such as cardiovascular and metabolic risk factors, and triglyceride and glucose concentrations[67].
All mutations associated with atherosclerosis are localized in the coding region of the mitochondrial genome. Mutations in two tRNALeu encoding genes affect the transport of leucine and can further inhibit the synthesis of respiratory chain enzymes and proteins[68,75]. The m.15059G>A mutation results in the formation of a stop codon. This formation stops the synthesis of cytochrome B, leading to the loss of 244 amino acids at the C-terminal of the protein, thus reducing its enzymatic function[68,75]. The m.652delG mutation might affect 12S rRNA subunit structure, leading to the disruption of mitochondrial ribosome function and subsequently causing a decrease in mitochondrial protein synthesis. In addition, mutations in mitochondrial NADH dehydrogenase subunit genes may cause defects in these enzymes and decreased cellular energy production[75]. Some of the mtDNA mutations can also alter monocyte/macrophage activation in atherosclerotic lesions through mitochondrial dysfunction[79]. The homoplasmic mtDNA mutations m.1811A>G and m.9477G>A (MT-CO3) are correlated with monocyte activation levels, whereas the heteroplasmic mutations m.14459G>A, m.1555A>G, and m.12315G>A are associated with
Mutations of mtDNA associated with cardiovascular disease
| Mutation | Gene | Associated processes and diseases | References |
| m.5592A>G | tRNAAla (MT-TA) | Alteration of tRNAAla and tRNAThr structures and function leading to mitochondrial dysfunction and a long-term increase in ROS generation in cardiovascular cells. CHD development. | [62] |
| m.15927G>A | tRNAThr (MT-TT) | ||
| m.8326A>G m.8331A>G m.8324T>A m.8344A>G | tRNALys (MT-TK) | Reduced availability of functional tRNALys leading to impairment of protein synthesis. Triple vessel disease, myocardial ischemia and CHD development. | [63] |
| m.8231C>A | Cytochrome c oxidase II | Cytochrome c oxidase deficiency and subsequently mitochondrial complex IV deficiency. CHD development. | [61] |
| m.5711A>G m.5725T>G | tRNAAsn (MT-TN) | Alteration of tRNAAsn and tRNATrp structures and function. CHD and coronary atherosclerosis development. | [64] |
| m.5568A>G | tRNATrp (MT-TW) | ||
| m.3970C>T m.4048G>A m.4071C>T m.4086C>T m.4164A>G m.4248 T>C | NADH dehydrogenase subunit I (MT-ND1) | Mutations in MT-ND1 associated with increased cytosolic levels of ROS. Mutations in MT-TQ associated with reduced levels of mitochondrial proteins. The mtDNA deletions disrupted most of the MT-ND2 genes. The mtDNA deletions may be an underlying cause of mitochondrial OXPHOS deficiency in post-mitotic cells. MIEH development. | [65] |
| m.4386T>C m.4394C>T | tRNAGln (MT-TQ) | ||
| m.4563delG m.4576delA m.4611delA m.4612delT | mtDNA 3777-4679 region | ||
| mtDNA4977 deletion | Five tRNA genes Four complex I subunits genes One complex IV subunit gene Two complex V subunits genes | Association with reductions in mtDNA-CN and respiration, promoting vascular smooth muscle cell and macrophage apoptosis as well as increased necrotic core and decreased fibrous cap areas. MACE, CHD, coronary atherosclerosis and breast cancer development. | [66] |
| m.15059G>A m.14846G>A | Cytochrome b | Mitochondria with structural defects were found in intimal cells carrying the mtDNA mutations in MT-RNR1, MT-TL1, MT-TL2 and MT-CYB genes. Mutation in MT-RNR1 affects protein synthesis. Mutations in MT-TL1 and MT-TL2 affect the transport of leucine and can further inhibit protein synthesis. Mutation m.15059G>A in MT-CYB leads to reduced enzymatic function of cytochrome B. Mutation m.14846G>A in MT-CYB, probably, stabilizes the complex of the III respiratory chain and increases the synthesis of ATP in the cell. Mutations in MT-RNR2 and MT-CO3 correlate with the levels of monocyte activation. Mutation in MT-ND1 is a silent mutation and does not cause the replacement of the amino acid in the NADH dehydrogenase subunit I. Probably, this mutation is linked to an atherogenic haplotype. Mutation in MT-ND2 results in the replacement of an amino acid in NADH dehydrogenase subunit II and is associated with atherosclerotic lesions. Mutation in MT-ND5 leads to the improved efficiency of complex 1 of the respiratory chain, leading to an increase in the production of energy in the cell. Mutation in MT-ND6 causes a defect in the enzyme, leading to the appearance of atherosclerotic lesions.Mutations MT-ND6, MT-RNR1 and MT-TL2 are associated with pro-inflammatory activation of monocytes. Atherosclerosis development. | [7,15,68-71,76-79] |
| m.12315G>A | tRNALeu (MT-TL2) | ||
| m.5178C>A | NADH dehydrogenase subunit II | ||
| m.3256C>T | tRNALeu (MT-TL1) | ||
| m.652delG | rRNA 12S | ||
| m.1555A>G | |||
| m.3336T>C | NADH dehydrogenase subunit I (MT-ND1) | ||
| m.14459G>A | NADH dehydrogenase subunit VI | ||
| m.13513G>A | NADH dehydrogenase subunit V | ||
| m.1811A>G | 16S RNA | ||
| m.9477G>A | Cytochrome c oxidase III | ||
| m.5024C>T | tRNAAla (MT-TA) | Associated with lower steady-state levels of tRNAAla and problems with intramitochondrial translation. Cardiomyopathies. | [80] |
Therefore, the combination of some of these mutations in the arterial intima may contribute to the formation of conditions that stimulate the development of atherosclerotic lesions and thickening of the intima-media layer of human arteries.
APPROACHES OF MITOCHONDRIAL DNA EDITING
Increased mutation levels are directly or indirectly involved in the development of atherosclerosis and other cardiovascular diseases, which suggests the possibility of gene therapy. There are various mtDNA editing technologies, such as mitochondrially targeted zinc-finger nucleases (mtZFNs), mitochondrial transcription activator-like effector nucleases (mitoTALENs), and technology based on clustered regularly interspaced short palindromic repeats (CRISPR) designed for specific cleavage of mtDNA sequences
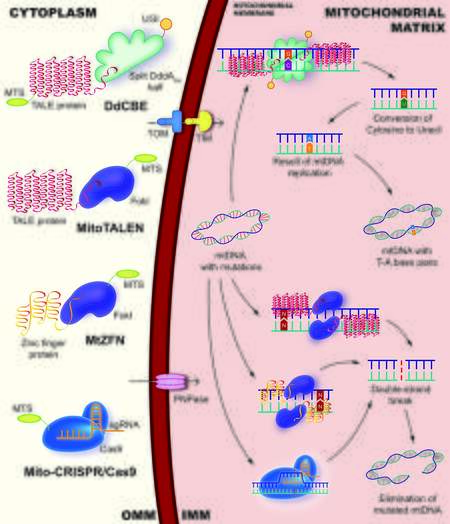
Figure 2. Main approaches of mitochondrial DNA editing. The DdCBE is able to catalyze the transition mutation of
Advantages and disadvantages of mtDNA editing technologies in mammalian cells
| Gene-editing technology | Advantages | Disadvantages | Application in cell and animal models |
| mtZFN | High specificity for mutant mtDNA RNA-free programmable dsDNA-binding protein ZFN Applicable to different cell types and species | Possible off-target activity but less than mito-CRISPR/Cas9 Relatively large size Acting as heterodimers Difficulties in packaging their coding genes into viral vectors Unable to recognize changes of individual bases in the genome sequence Requires repeated transfection | Neuropathy ataxia retinitis pigmentosa syndrome, cardiomyopathies |
| mitoTALEN | High specificity for mutant mtDNA RNA-free programmable dsDNA-binding protein TALEN Applicable to different cell types and species | Cardiomyopathies, mitochondrial diseases myoclonic epilepsy with ragged red fibers, MELAS/Leigh syndrome | |
| mitoARCUS | Monomeric High specificity for mutant mtDNA Smaller size compared to mitoTALEN and mtZFN Recognize sequences that differ by only one base pair Applicable to different cell types and species | Possible off-target activity but less than mito-CRISPR/Cas9 Commercial technology that has not received wide distribution | Cardiomyopathies |
| mito-CRISPR/Cas9 | High specificity for mutant mtDNA Easy to design Many CRISPR-associated protein nucleases can be used Can be delivered inside the mitochondrial matrix as a CRISPR-RNP complex Applicable to different cell types and species | More off-target activity than other editing technologies Requires complex delivery systems Lack of effective methods for delivering sgRNA through the mitochondrial membrane Poorly understood mtDNA repair mechanisms Temperature difference in the nucleus and mitochondria may affect the efficiency and specificity of Cas9 | MELAS, Kearns-Sayre syndrome, Atherosclerosis |
| DdCBE | Precise editing of mtDNA mutations CRISPR-free RNA-free programmable dsDNA-binding protein TALEN Lack of requirement for a PAM Wide editing window, that can lead to deamination of several cytidines simultaneously on both DNA strands DSB not used Applicable to different cell types and species | Possible off-target activity but less than other editing technologies | MELAS/Leigh syndrome, LHON |
MtZFN is a chimeric enzyme, which includes Cys2His2 zinc-finger protein conjugated to the C-terminal FokI catalytic enzyme and domain with an additional mitochondrial targeting sequence (MTS) and nuclear export signal peptides that provide mitochondrial localization[83]. Recently, mtZFN has been shown to be capable of successfully shifting mtDNA heteroplasmy in model cybrid cells[83]. These cybrid cells carry the m.8993T>G mutation associated with neuropathy ataxia retinitis pigmentosa syndrome, which results in aL217R replacement in a highly conserved residue of subunit a of ATP synthase. Additionally, mtZFNs were shown to successfully eliminate the m.5024C>T mutation in the tRNAAla gene in cardiac muscle tissue in a mouse model[80]. The m.5024C>T mutation is associated with tRNAAla instability, myopathies, cardiomyopathies, and oxidative phosphorylation deficiency.
MitoTALENs consist of a DNA-binding domain fused to the FokI endonuclease domain and mtZFN, and function as dimers[82]. Each mtDNA targeting mitoTALEN monomers contains 14.5-16.5 repeats. One of the monomers targets the mutation sequence, and the other monomer binds to the wild-type sequence with a spacer length of 14-17 bp. This determines the specific cleavage of the mutant mtDNA upon dimerization of the FokI nuclease.
A decreased mutation load in skeletal muscles and the heart was successfully demonstrated using mitoTALENs in the mouse model with the m.5024C>T heteroplasmic mutation in the tRNAAla gene[84]. A vector based on the adeno-associated virus 9 was used as a delivery method. A single injection of adeno-associated virus 9-mitoTALEN had a long and stable effect for up to 10-24 weeks.
The mitoTALENs can eliminate mutant mtDNA from transmitochondrial cybrid cells containing m.8344A>G mutations in the tRNALys gene and m.13513G>A mutations in the MT-ND5 gene associated with myoclonic epilepsy with ragged red fibers and MELAS/Leigh syndrome, respectively[85]. The application of mitoTALENs to cybrid cells efficiently reduces levels of the targeted mutant mtDNAs. This application also restores respiratory function of the cells and the activity of oxidative phosphorylation enzymes. In addition, decreased heteroplasmy levels of MELAS-associated mutations, such as m.3243A>G and m.13513G>A, were demonstrated by several studies that applied mitoTALENs to induced pluripotent stem cells[82,86].
Molecular hybrids, such as I-TevI-TALE (mitoTev-TALE), were developed further for better efficiency of gene-editing technologies based on TALEN. MitoTev-TALEs are monomeric GIY-YIG homing nucleases from a T4 phage (I-TevI) associated with the DNA-binding domain of TALE[87]. Such a modification can facilitate the delivery of the editing systems to the damaged tissue for gene therapy. These mitoTev-TALEs successfully decrease heteroplasmy levels of the m.8344A>G mutation in mtDNA, resulting in improved oxidative phosphorylation function of cells carrying the mutation.
As an alternative to mitoTALEN/ZFN restriction endonucleases, the editing system mitoARCUS based on the homing endonuclease I-CreI has recently been created[88]. In contrast to the technologies mentioned above, I-CreI is monomeric, has a relatively small size, and is able to recognize sequences that differ by only one bp. I-CreI was first discovered in Chlamydomonas reinhardtii chloroplasts. I-CreI is a member of the meganuclease family with the LAGLIDADG22 motif, and is a homodimeric enzyme converted to a monomer using a small peptide linker (1092 bp/40 kDa). This gene-editing system binds to a palindromic double-stranded DNA (dsDNA) sequence of 22 bp, causing the formation of DSBs[88]. Additionally, mitoARCUS can decrease heteroplasmy levels up to 60% for 24 hours after transfection, and maintain this level for up to 3 weeks, which indicates a high specificity for mutant mtDNA. A previous study used mitoARCUS in vivo in mice intravenously[88]. This study showed an effective shift in heteroplasmy levels of the m.5024C>T mutation in the tRNAAla gene in liver and skeletal muscles of mice without depletion of total mtDNA levels after 6, 12, or 24 weeks[88].
Taken together, mitoTALEN/ZFN studies have clearly shown that such gene-editing technologies can be effective tools in in vivo and in vitro experiments without exhibiting high cytotoxicity in cells and tissues[80,84]. The possibility of application of mitoTALEN/ZFN to cybrid cell lines containing mutations in mtDNA associated with atherosclerosis is of particular interest for studying the cellular mechanisms of atherogenesis. An example of such a study is that mitoTALEN/ZFN could be used in recently created cybrid cell lines carrying the m.12315G>A mutation in the tRNALeu gene[89]. Because these gene-editing technologies were previously used to decrease heteroplasmy levels of mutations in mitochondrial tRNA genes, they can be adapted to the cybrid cell model mentioned above.
Another mtDNA heteroplasmy editing and decreasing technology is the CRISPR-based approach, which is widely used for nuclear genome editing[50,54,90,91]. CRISPRs are part of the bacterial immune system and recognize the protospacer adjacent motif (PAM). The PAM is a two to six-bp DNA sequence following the DNA sequence targeted by the endonuclease[92]. The mito-CRISPR mitochondrial editing system includes CRISPR-associated protein-9 nuclease (Cas9) or CRISPR-associated protein 12a (Cas12a), also known as Cpf1, as well as a chimeric single-guide RNA (sgRNA) that recognizes the PAM sequence[93]. The efficiency of mito-CRISPR systems depends on aspects, such as the targeted delivery of CRISPR nuclease to the mitochondria by fusion with MTS and mitochondrial targeting using sgRNA. Additionally, this efficiency depends on the formation of a functional CRISPR ribonucleoprotein (RNP) complex consisting of Cas9 protein and sgRNA inside the mitochondrial matrix, as well as the functional nuclease activity of the CRISPR-RNP complex[49,93].
Studies have shown the effectiveness of the mito-CRISPR/Cas9 gene-editing system in reducing mutant mtDNA in cells[93]. Mitochondrial targeting efficiency and effects on mitochondrial dynamics/function have been demonstrated by using several CRISPR nucleases. These nucleases include SpCas9 type II
Recent studies on the mito-CRISPR/Cas9 system based on the pSpCas9-mito vector have also shown the ability to reduce the number of mtDNA copies in eukaryotic cells and zebrafish (Danio rerio)[50]. This gene editing system included two flanking MTSs to COX8A (cytochrome c oxidase subunit 8A). Additionally, MT-ND1 and MT-ND4 (NADH dehydrogenase subunit IV) genes, as well as two mitochondrial sites of the D-loop in Danio rerio were used as target sites. Furthermore, mito-CRISPR/Cas9-damaged mtDNA is capable of being repaired by exogenous single-stranded DNA (ssDNA) (Oligo-HEX) via the HR mechanism in human HEK-293T and Danio rerio mitochondria. The findings of this study are consistent with those of the ability of sgRNA to be imported into mitochondria, and are supported by data on the possibility of import and colocalization of ssDNA and mitoCas9 with mtDNA[91].
To date, the most promising method to deliver editing systems based on CRISPR/Cas9 to mitochondria appears to be the natural RNA import pathway into organelles. The delivery of recombinant RNA complementary to the mutant mtDNA sequence into mitochondria leads to the inhibition of mtDNA replication followed by decreased heteroplasmy levels[94]. To create a mutant mtDNA editing approach on the basis of a mitochondrial RNA import pathway, an attempt was made to create a specific CRISPR/Cas9 system targeting mtDNA. A study suggested that the number of mtDNA copies decreased by two to three times[91]. In this study, the editing system was a mitochondrially targeted Cas9 (MTSCOX8A-hCas9), and a set of two sgRNAs targeting the non-coding region and the mtDNA CytB gene (MT-CYB) sequence[91]. This study suggests that binding of the hCas9/sgRNA complex in the non-coding region results in the disruption of D-loop formation and mtDNA replication, thus making other mtDNA regions more accessible for cleavage by the second hCas9/sgRNA complex.
Application of the CRISPR/Cas9 system is also possible for removing somatic mutations in the mitochondrial genome of cells in atherosclerotic lesions. According to previous studies, pMitoCas9 reduces the level of mutant mtDNA in THP-1 cybrid cells carrying the m.15059G>A mutation in the MT-CYB gene associated with atherosclerosis[22-24,68]. The cybrid cell line used in these studies was created by fusion of the mtDNA-depleted human monocyte-like cell line THP-1 and mitochondria from donor platelets with high heteroplasmy levels of the mutations associated with atherosclerosis[89,95]. These cybrid cells can be used to model the occurrence and development of atherosclerosis in cells. These cells can also be used to assess the changes occurring in immune cells owing to the presence of mutations in their mtDNA. The mito-CRISPR/Cas9 designed in these studies can be applied not only to model cell lines, but also to aortic intimal cells isolated from donors with identified specific mtDNA mutations associated with atherosclerosis and cardiovascular disease[22-24,68]. Such cells can be resident smooth muscle cells, pericyte-like cells, and endothelial cells, which are major participants in the development of atherosclerotic lesions[96-98]. However, the technology for eliminating mutant mtDNA and for targeted delivery of editing systems to tissues and cells is currently not well optimized. Therefore, more in-depth research is required in this area. Nevertheless, mito-CRISPR/Cas9 appears to be a promising technology for studying the role of mutations associated with cellular mechanisms of human atherogenesis.
Existing approaches of mtDNA editing, such as mito-CRISPR/Cas9, mtZFN, and mitoTALEN technologies, remain difficult to implement, despite their prospects for application. There is growing evidence on the application of editing systems based on bacterial toxins, which include a group of toxins of the deaminase superfamily catalyzing the deamination of ssDNA, RNA, free nucleosides, nitrogenous bases, and other nucleotide derivatives[43,44]. To perform bp editing on dsDNA, cytosine base editors (CBEs) require the CRISPR-Cas9 system to unwind the DNA double helix. However, recent studies in the field of editing the mitochondrial genome of eukaryotic cells provide an opportunity to perform CBEs without CRISPR-based systems[43]. Therefore, the effectiveness of RNA-free deoxycytidine deamination-derived cytosine base editors (DdCBEs) was demonstrated. DdCBEs are able to catalyze the transition mutation of C•G base pairs to T•A base pairs by deamination of deoxycytidine to deoxyuridine as a mutagenic intermediate.
In contrast to existing CRISPR-based cytosine base editors, DdCBEs consist of two-halves of an interbacterial toxin that catalyzes the deamination of cytidines within dsDNA (DddAtox). DdCBEs are activated by assembling together on the target DNA, as well as by transcription activator-like effector (TALE) proteins and uracil glycosylase inhibitors[43,99]. The application of RNA-free programmable dsDNA-binding proteins, such as ZFN or TALE, allows for an increase in the accuracy of targeting DddAtox to mtDNA without applying CRISPR and sgRNA. DddAtox has a much wider editing window than cytosine base editors of 14-18 bp, which can lead to the deamination of several cytidines simultaneously on both DNA strands[44]. CRISPR-free DdCBE systems allow the precise editing of mtDNA mutations without causing DSB formation and a decrease in mtDNA copies. This could be important in the study and therapy of mitochondrial diseases.
The effectiveness of DdCBEs in editing mtDNA bp in HEK293T cells can be up to 49% on days 3-6, depending on the type and orientation of the cleavage, and the target position of cytosine in the spacing region[43]. The stability of mtDNA editing in HEK293T cells for 18 days has successfully been shown. However, this stability does not lead to a decrease in cell viability or the presence of large deletions in mtDNA, and has no effect on the number of mtDNA copies. Moreover, the effectiveness of DdCBEs was successfully shown by editing the MT-ND4 gene in cells containing the m.11922G>A mutation.
The application of DdCBEs is possible not only in immortalized eukaryotic cell lines, but also in mouse and Danio rerio embryos[99,100]. Recently, an attempt was made to create a mouse model of mitochondrial diseases by applying DdCBEs to C57BL6/J mouse embryos[99]. The mitochondrial MT-ND5 gene was chosen as the target gene for editing to obtain the m.12918G>A mutation. This mutation in humans is associated with MELAS and some symptoms of Leigh syndrome and LHON[2]. The application of DdCBEs induces a shift in mtDNA heteroplasmy in zygotes at the unicellular stage, with an editing efficiency of 0.25%-23% maintained throughout the development and differentiation of the embryo. Newborn mice developed from embryos carry the mutant allele with a frequency of 3.9%-31.6%[99]. A similar application of DdCBEs to create zebrafish models for mitochondrial diseases was also successful. A study showed achievement of editing efficiency of up to 67.9% for the introduction of the D393N mutation for the MT-ND5 gene associated with Leigh syndrome and MELAS[100].
These findings mentioned above strongly suggest that DdCBEs can be used for editing mtDNA point mutations and for creating animal models of diseases associated with mutations in the mitochondrial genome. This editing technology could be a promising approach for the creation of cell or animal models containing mutations, such as m.15059G>A, m.14846G>A, m.12315G>A, m.3256C>T, m.14459G>A, m.13513G>A, and m.9477G>A, which are associated with atherosclerosis. Unlike existing cybrid cell models, animal models with such mutations in mtDNA are useful in studying the pathogenesis of atherosclerosis and pathological processes occurring in organs and tissues. In addition, DdCBEs could be used for editing mutations, such as m.1555A>G, m.3336T>C, and m.1811A>G, by C•G to T•A bp conversions. Such a highly effective editing technology would allow the removal of mutations in mtDNA with great accuracy without DSB formation followed by the elimination of mtDNA. Such abilities of DdCBEs may be an advantage in studying the role of individual mutations in atherogenesis, as well as in the further development of approaches aimed at treating this disease.
CHALLENGES OF MITOCHONDRIAL GENOME EDITING
Despite the success achieved in the application of restriction endonucleases, researchers are faced with a number of limitations arising from the structural features of mitochondria and the components of editing systems [Table 2]. Although mtZFNs and mitoTALENs have been successful in shifting mtDNA heteroplasmy, these editing systems have a number of disadvantages that may impede the study of the mitochondrial genome. The main challenges include the relatively large size of gene-editing systems and their acting as heterodimers. These challenges lead to difficulties in packaging their coding genes into viral vectors. Therefore, selecting the most compatible delivery systems into cells is necessary. In addition, mtZFNs and mitoTALENs are unable to recognize changes in individual bases in the genome sequence, and their application requires repeated transfection of cells to achieve an effective decrease in mtDNA heteroplasmy levels[81,85]. The recently created mitoARCUS could be more successful and widely used than mitoTALEN/ZFN because of its smaller size. This smaller size allows for the packaging and delivery of a single recombinant adeno-associated virus with a packaging limit of up to 4.5 kb[88]. However, currently, mitoARCUS is a commercial project that has not received wide distribution. A promising competitor to mitoTALEN/ZFN may be the DdCBE system.
One of the main problems in the application of mito-CRISPR/Cas9 is the insufficiently explored pathways for the delivery of nucleic acids to mitochondria[101]. A particular difficulty in the development of CRISPR/Cas9-mediated gene editing of mtDNA is the lack of effective methods for delivering sgRNA through the mitochondrial membrane. The structure and features of mitochondrial transport may not allow most nucleic acids to transport into the organelles. Therefore, mitochondria are poorly accessible for gene-editing systems based on CRISPR. Furthermore, the difference in mitochondrial import efficiency observed by applying different nucleases could be explained by different characteristics of proteins, such as the organization of the enzyme domain and N-terminal secondary structures, and differences in the total peptide charge of imported proteins[93]. An example of these differences is that LbCas12a is smaller, more positively charged, and less hydrophobic than SpCas9, which may explain its higher mitochondrial targeting efficiency.
To solve the problem associated with the transport of sgRNA and Cas9 into mitochondria, the polynucleotide phosphorylase protein encoded by the PNPT1 gene may be applied[90]. This transport protein is present in the inner membrane and intermembrane space of mitochondria, and carries out the transfer of rRNA, tRNA, and microRNA[102]. Another pathway for importing gene-editing systems into mitochondria may be transport through the translocase of the outer mitochondrial membrane (TOM)/translocase of the inner mitochondrial membrane (TIM) complex, which is a mitochondrial protein-import machinery[103]. The use of this pathway was shown in a study of applying LbCas12a and mitochondrial import blockers[93]. The localization and expression of LbCas12a was successfully demonstrated in the mitochondrial matrix in wild-type cells and in MELAS cybrids. However, to be able to import gene-editing systems by TOM/TIM, differences in the total peptide charge of imported proteins, which stimulate mitochondrial import, need to be taken into account.
The application of a canonical mitochondrial localization signal or MTS has been successfully shown for targeted transport of editing systems into the mitochondrial matrix[83,90,104]. MTSs are short peptides of 15-70 amino acids long, which carry positively charged basic residues. The charge, length, and structure of the MTS must be taken into account for the import of proteins into mitochondria. The most common MTS sites used in studies are the SOD2 and COX8A gene sequences[50,85,91]. A hybrid sgRNA was developed that is specific to the 11205G mutant region in the MT-ND4 gene and includes a 20-nucleotide stem-loop element, which is a component of nuclear RNAse P, at the 5′ end of the guide sequence[90]. Cell transfection using sgRNA together with mitochondria-targeting Cas9 constructs (mitochondrial localization signal-Cas9) results in colocalization of the sgRNA sequence with the RP-loop with mitochondria and a considerable decrease in the mtDNA level carrying the mutation.
An alternative approach for mitochondrial gene delivery is liposomes. Liposomes can be considerably modified to reduce cytotoxicity and increase the selectivity of the delivered DNA or RNA[105,106]. Therefore, important features for mitochondrial liposomes may be the composition of membrane lipids, the relevant molar ratio of liposome components, size, molecular weight, and the ratio of positively charged polymer amine (N) groups to negatively charged nucleic acid phosphate (P) groups (N/P), which determine the charge of the liposomes. Recently, a liposome-based mitochondrial delivery system called MITO-Porter was developed for the delivery of encapsulated substances to mitochondria via membrane fusion[106,107]. However, such a delivery system is not universal and requires optimization for specific use with cell lines[108].
Another method to solve the challenge of CRISPR system transport into mitochondria is Edit Plasmids, which includes a mitochondrial codon-optimized Cas9 expression cassette, an sgRNA expression cassette, donor DNA for integration between two DSB sites induced by the Cas9/sgRNA complex, and a selectable or screening marker[49]. The insertion of donor DNA into target sites, and maintenance and autonomous replication of Edit Plasmids in mitochondria for a few dozens of generations in the presence of the wild-type genome has been demonstrated.
Furthermore, the existence of mtDNA repair mechanisms in mitochondria is one of the challenges in mitochondrial genome editing. As mentioned above, homologous recombination and MMEJ repair mechanisms may occur in mitochondria in addition to the BER mechanism[43,44,46-54]. MMEJ leads to the formation of small InDel mutations in flanking DNA segments or major deletions in the sequence[109]. InDel formation is a mutagenic process that can negatively affect the result of genome editing. Moreover, DNA repair mechanisms require the presence of an ssDNA or dsDNA for the repair of DSBs forming as a result of the action of restriction endonucleases. A recent study on the application of mito-CRISPR/Cas9 and Oligo-HEX ssDNA showed that the sequence of ssDNA is inserted into the sites of DSB formation[50]. A possible solution to this challenge could be inhibitors of proteins that mediate the repair mechanisms. However, the issue of specific proteins of MMEJ and homologous recombination in mitochondria and the consequences of their inhibition remain poorly examined.
The possibility of off-target activity in the nuclear genome should also be considered. Off-target activity can occur during the application of mitochondria-targeted editing systems when they enter the cell nucleus. The gene-editing system needs to target a mtDNA sequence with high specificity to successfully decrease the heteroplasmy level. In a previous study that compared the effectiveness of CRISPR nucleases, the authors noted that off-target cleavage of nuclear DNA may be a serious problem for LbCas12a[93,110]. The studies mentioned above suggest that proper selection of nucleases and approaches to targeting gene-editing systems to mtDNA is extremely important.
The off-target activity in the mitochondrial genome has been studied using promising DdCBE editing systems[43,111]. A high average frequency of off-target editing may occur if DdCBEs contain a permissive mutant N-terminal domain of TALE, which can increase the non-specific binding of TALE arrays. Additionally, DdCBEs with the standard N-terminal domain show 150-860 times more on-target editing[43]. DdCBEs halves containing TALE arrays with more non-specific DNA-binding activity bind proximally to temporarily reassemble active DddAtox, which can then involve non-target mtDNA regions. However, the frequency of mitochondrial off-target activity remains relatively low[111]. The development of in vitro and
Special attention should be paid to the temperature difference in the nucleus and mitochondria in case of editing the mitochondrial genome by systems based on CRISPR/Cas9. Cas9 nucleases are temperature-sensitive, and may affect the efficiency and specificity of their ability to cleave DNA[112]. According to an existing hypothesis, the temperature in mitochondria can be 10 °C higher than that in the cytosol and nucleus of the cell, and be approximately 48-50 °C[113]. Such a shift in the temperature is likely to affect the function of the RNP complex. Moreover, an increase in the temperature by 2 °C can enhance the efficiency of CRISPR/Cas9 as an RNP complex during nuclear genome editing in some cell lines, but at the same time, even that small increase enhances off-target activity[112]. Because of these features, the specificity of CRISPR/Cas9 in mitochondria can be improved by creating specific sgRNAs with a minimum potential level of off-target activity or by applying other modified thermostable Cas9 variants with higher accuracy[112,114]. Notably, in studies that aimed to decrease the level of mtDNA heteroplasmy by recombinant RNA, RNA with higher melting points proved to be the most effective[115]. It may also indicate an existing problem of temperature differences in mitochondria and other parts of the cell. Therefore, the intracellular temperature gradient should be taken into account for developing mitochondria-targeted editing systems.
CONCLUSION
Mutations in mtDNA not only cause many hereditary human diseases, but also contribute to the complication and development of non-hereditary diseases such as atherosclerosis. Despite some progress in the development of approaches to mitochondrial genome editing, such as mtZFN, mitoTALEN, and mito-CRISPR/Cas9, the possibilities of their application for therapy and treatment of diseases caused by mtDNA mutations remain limited. Further development and optimization of methods for manipulating the mitochondrial genome, taking into account the emerging problems associated with the delivery of editing constructs and the possible occurrence of off-target activity, may provide a potential treatment for diseases caused by mtDNA mutations in the future.
DECLARATIONS
Authors’ contributionsConceptualized the manuscript: Khotina VA, Sukhorukov VN
Wrote the manuscript text and made a visualization: Khotina VA
Reviewed the text: Bagheri Ekta M, Baig MS, Wu WK, Grechko AV
Developed the methodology: Bagheri Ekta M, Sukhorukov VN
Completed the formal analysis: Baig MS, Wu WK
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the Ministry of Science and Higher Education of the Russian Federation, Project «Age-related changes in the regulation of vascular and myocardial contractility, mechanisms for the development of vascular pathology», N° FGFU-2022-0008.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. López-Gallardo E, Cammarata-Scalisi F, Emperador S, et al. Mitochondrial DNA pathogenic mutations in multiple symmetric lipomatosis. Clin Genet 2020;97:731-5.
2. Shanske S, Coku J, Lu J, et al. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: Evidence from 12 cases. Arch Neurol 2008;65:368-72.
3. Bargiela D, Chinnery PF. Mitochondria in neuroinflammation - multiple sclerosis (MS), leber hereditary optic neuropathy (LHON) and LHON-MS. Neurosci Lett 2019;710:132932.
4. Lorenz R, Ahting U, Betzler C, Heimering S, Borggräfe I, Lange-Sperandio B. Homoplasmy of the mitochondrial DNA mutation m.616T>C leads to mitochondrial tubulointerstitial kidney disease and encephalopathia. Nephron 2020;144:156-60.
5. Decoux-Poullot AG, Bannwarth S, Procaccio V, et al. Clinical phenotype of mitochondrial diabetes due to rare mitochondrial DNA mutations. Ann Endocrinol 2020;81:68-77.
6. Hu H, Lin Y, Xu X, Lin S, Chen X, Wang S. The alterations of mitochondrial DNA in coronary heart disease. Exp Mol Pathol 2020;114:104412.
7. Sobenin IA, Sazonova MA, Postnov AY, Bobryshev YV, Orekhov AN. Mitochondrial mutations are associated with atherosclerotic lesions in the human aorta. Clin Dev Immunol 2012;2012:832464.
8. Man JJ, Beckman JA, Jaffe IZ. Sex as a biological variable in atherosclerosis. Circ Res 2020;126:1297-319.
9. Bäck M, Yurdagul A, Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 2019;16:389-406.
10. Ito F, Sono Y, Ito T. Measurement and clinical significance of lipid peroxidation as a biomarker of oxidative stress: oxidative stress in diabetes, atherosclerosis, and chronic inflammation. Antioxidants 2019;8:72.
11. Soldatov VO, Malorodova TN, Balamutova TI, Ksenofontov AO, Dovgan AP, Urozhevskaya ZS. Endothelial dysfunction: comparative evaluation of ultrasound dopplerography, laser dopplerflowmetry and direct monitoring of arterial pressure for conducting pharmacological tests in rats. Res Results Pharmacol 2018;4:73-80.
12. Soldatov VO, Malorodova TN, Pokrovskaya TG, et al. Ultrasonic dopplerography for the evaluation of endothelial function in the conduct of pharmacological vascular samples in an experiment. Int J Res Pharm Sci 2018;9:735-40.
13. Puchenkova OA, Nadezhdin SV, Soldatov VO, et al. Study of antiatherosclerotic and endothelioprotective activity of peptide agonists of EPOR/CD131 heteroreceptor. Farm Farmakol 2020;8:100-11.
14. Glanz VY, Sobenin IA, Grechko AV, Yet SF, Orekhov AN. The role of mitochondria in cardiovascular diseases related to atherosclerosis. Front Biosci 2020;12:102-12.
15. Sazonova MA, Sinyov VV, Ryzhkova AI, et al. Role of mitochondrial genome mutations in pathogenesis of carotid atherosclerosis. Oxid Med Cell Longev 2017;2017:6934394.
16. Mercer JR, Cheng KK, Figg N, et al. DNA Damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ Res 2010;107:1021-31.
17. Quan Y, Xin Y, Tian G, Zhou J, Liu X. Mitochondrial ROS-modulated mtDNA: a potential target for cardiac aging. Oxid Med Cell Longev 2020;2020:9423593.
18. Arauna D, Furrianca M, Espinosa-Parrilla Y, Fuentes E, Alarcón M, Palomo I. Natural bioactive compounds as protectors of mitochondrial dysfunction in cardiovascular diseases and aging. Molecules 2019;24:4259.
19. Kornfeld OS, Hwang S, Disatnik MH, Chen CH, Qvit N, Mochly-Rosen D. Mitochondrial reactive oxygen species at the heart of the matter: new therapeutic approaches for cardiovascular diseases. Circ Res 2015;116:1783-99.
20. Ortiz JM, Swerdlow RH. Mitochondrial dysfunction in Alzheimer’s disease: role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol 2019;176:3489-507.
21. Gutierrez-Mariscal FM, De Larriva APA, Limia-Perez L, Romero-Cabrera JL, Yubero-Serrano EM, López-Miranda J. Coenzyme Q10 supplementation for the reduction of oxidative stress: clinical implications in the treatment of chronic diseases. Int J Mol Sci 2020;21:7870.
22. Sukhorukov VN, Kalmykov VA, Khotina VA, Sinyov VV, Khasanova ZB, Sobenin IA. Approach to edit mitochondrial DNA mutations associated with atherosclerosis. Atherosclerosis 2021;331:70-1.
23. Sukhorukov VN, Kalmykov VA, Khotina VA, et al. Elimination of atherosclerosis related mutation from mitochondrial cytb gene. In proceedings of the 19th international symposium atheroscler, Kyoto, Japan, 24-27 Oct 2021.
24. Sukhorukov VN, Kalmykov VA, Khotina VA, Omelchenko AV, Orekhova VA, Orekhov AN. Mitochondrial DNA CRISPR/Cas9 editing: an approach to establishing the role of mitochondrial mutations in atherogenesis. In proceedings of the 90th European atherosclerosis society congress (EAS2022), Milan, Italy, 22-25 May 2022.
25. Kastaniotis AJ, Autio KJ, Kerätär JM, et al. Mitochondrial fatty acid synthesis, fatty acids and mitochondrial physiology. Biochim Biophys Acta Mol Cell Biol Lipids 2017;1862:39-48.
26. Song J, Herrmann JM, Becker T. Quality control of the mitochondrial proteome. Nat Rev Mol Cell Biol 2021;22:54-70.
27. Abate M, Festa A, Falco M, et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin Cell Dev Biol 2020;98:139-53.
29. Faas MM, de Vos P. Mitochondrial function in immune cells in health and disease. Biochim Biophys Acta Mol Basis Dis 2020;1866:165845.
31. Protasoni M, Zeviani M. Mitochondrial structure and bioenergetics in normal and disease conditions. Int J Mol Sci 2021;22:586.
32. Nicholls TJ, Gustafsson CM. Separating and segregating the human mitochondrial genome. Trends Biochem Sci 2018;43:869-81.
33. Nicholls TJ, Minczuk M. In D-loop: 40 years of mitochondrial 7S DNA. Exp Gerontol 2014;56:175-81.
34. Omasanggar R, Yu CY, Ang GY, et al. Mitochondrial DNA mutations in malaysian female breast cancer patients. PLoS One 2020;15:e0233461.
35. Rodrigues SC, Cardoso RMS, Duarte FV. Mitochondrial microRNAs: a putative role in tissue regeneration. Biology 2020;9:486.
36. Ott M, Amunts A, Brown A. Organization and regulation of mitochondrial protein synthesis. Annu Rev Biochem 2016;85:77-101.
37. Kukat C, Davies KM, Wurm CA, et al. Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc Natl Acad Sci USA 2015;112:11288-93.
38. Gustafsson CM, Falkenberg M, Larsson NG. Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem 2016;85:133-60.
39. Markin AM, Khotina VA, Zabudskaya XG, et al. Disturbance of mitochondrial dynamics and mitochondrial therapies in atherosclerosis. Life 2021;11:165.
40. Falkenberg M. Mitochondrial DNA replication in mammalian cells: overview of the pathway. Essays Biochem 2018;62:287-96.
41. Fontana GA, Gahlon HL. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res 2020;48:11244-58.
42. Yasukawa T, Kang D. An overview of mammalian mitochondrial DNA replication mechanisms. J Biochem 2018;164:183-93.
43. Mok BY, de Moraes MH, Zeng J, et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020;583:631-7.
44. Gu S, Bodai Z, Cowan QT, Komor AC. Base editors: expanding the types of DNA damage products harnessed for genome editing. Gene Genome Ed 2021;1:100005.
45. Kamenisch Y, Fousteri M, Knoch J, et al. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J Exp Med 2010;207:379-90.
46. Stein A, Kalifa L, Sia EA. Members of the RAD52 epistasis group contribute to mitochondrial homologous recombination and double-strand break repair in saccharomyces cerevisiae. PLoS Genet 2015;11:e1005664.
47. Chesner LN, Essawy M, Warner C, Campbell C. DNA-protein crosslinks are repaired via homologous recombination in mammalian mitochondria. DNA Repair 2021;97:103026.
48. Mbantenkhu M, Wang X, Nardozzi JD, et al. Mgm101 is a Rad52-related protein required for mitochondrial DNA recombination. J Biol Chem 2011;286:42360-70.
49. Yoo BC, Yadav NS, Orozco EM, Sakai H. Cas9/gRNA-mediated genome editing of yeast mitochondria and Chlamydomonas chloroplasts. PeerJ 2020;8:e8362.
50. Bian WP, Chen YL, Luo JJ, Wang C, Xie SL, Pei DS. Knock-In strategy for editing human and zebrafish mitochondrial DNA using Mito-CRISPR/Cas9 system. ACS Synth Biol 2019;8:621-32.
51. Dahal S, Dubey S, Raghavan SC. Homologous recombination-mediated repair of DNA double-strand breaks operates in mammalian mitochondria. Cell Mol Life Sci 2018;75:1641-55.
52. Tadi SK, Sebastian R, Dahal S, Babu RK, Choudhary B, Raghavan SC. Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions. Mol Biol Cell 2016;27:223-35.
53. Reddy P, Ocampo A, Suzuki K, et al. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 2015;161:459-69.
54. Wang B, Lv X, Wang Y, et al. CRISPR/Cas9-mediated mutagenesis at microhomologous regions of human mitochondrial genome. Sci China Life Sci 2021;64:1463-72.
55. Wisnovsky S, Jean SR, Kelley SO. Mitochondrial DNA repair and replication proteins revealed by targeted chemical probes. Nat Chem Biol 2016;12:567-73.
56. de Souza-Pinto NC, Mason PA, Hashiguchi K, et al. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair 2009;8:704-19.
57. Mustafa MF, Fakurazi S, Abdullah MA, Maniam S. Pathogenic mitochondria DNA mutations: current detection tools and interventions. Genes 2020;11:192.
58. Myasoedova VA, Di Minno A, Songia P, et al. Sex-specific differences in age-related aortic valve calcium load: A systematic review and meta-analysis. Ageing Res Rev 2020;61:101077.
59. Sazonova M, Shkurat T, Demakova N, et al. Mitochondrial genome sequencing in atherosclerosis: what’s next? Curr Pharm Des 2016;22:390-6.
60. Gonzalez-Freire M, Moore AZ, Peterson CA, et al. Associations of peripheral artery disease with calf skeletal muscle mitochondrial dna heteroplasmy. J Am Heart Assoc 2020;9:e015197.
61. Heidari MM, Mirfakhradini FS, Tayefi F, Ghorbani S, Khatami M, Hadadzadeh M. Novel point mutations in mitochondrial MT-CO2 gene may be risk factors for coronary artery disease. Appl Biochem Biotechnol 2020;191:1326-39.
62. Qin Y, Xue L, Jiang P, et al. Mitochondrial tRNA variants in Chinese subjects with coronary heart disease. J Am Heart Assoc 2014;3:e000437.
63. Matam K, Shaik NA, Aggarwal S, et al. Evidence for the presence of somatic mitochondrial DNA mutations in right atrial appendage tissues of coronary artery disease patients. Mol Genet Genomics 2014;289:533-40.
64. Heidari MM, Derakhshani M, Sedighi F, Foruzan-Nia SK. Mutation analysis of the mitochondrial tRNA genes in Iranian coronary atherosclerosis patients. Iran J Public Health 2017;46:1379-85.
65. Zhu Y, You J, Xu C, Gu X. Associations of mitochondrial DNA 3777-4679 region mutations with maternally inherited essential hypertensive subjects in China. BMC Med Genet 2020;21:105.
66. Vecoli C, Borghini A, Pulignani S, et al. Prognostic value of mitochondrial DNA4977 deletion and mitochondrial DNA copy number in patients with stable coronary artery disease. Atherosclerosis 2018;276:91-7.
67. Sobenin IA, Salonen JT, Khasanova ZB, et al. Carotid atherosclerosis-related mutations of mitochondrial DNA do not explain the phenotype of metabolic syndrome. Vessel Plus 2019;3:14.
68. Sobenin IA, Sazonova MA, Postnov AY, Bobryshev YV, Orekhov AN. Changes of mitochondria in atherosclerosis: Possible determinant in the pathogenesis of the disease. Atherosclerosis 2013;227:283-8.
69. Kirichenko TV, Ragino YI, Voevoda MI, et al. Data on association of mitochondrial heteroplasmy with carotid intima-media thickness in subjects from Russian and Kazakh populations. Data Brief 2020;29:105136.
70. Sobenin IA, Myasoedova VA, Kirichenko TV, et al. Profiling of risk of subclinical atherosclerosis: Possible interplay of genetic and environmental factors as the update of conventional approach. Vessel Plus 2019;3:15.
71. Kirichenko TV, Ryzhkova AI, Sinyov VV, et al. Impact of mitochondrial dna mutations on carotid intima-media thickness in the Novosibirsk region. Life 2020;10:160.
72. Sinyov VV, Sazonova MA, Ryzhkova AI, et al. Potential use of buccal epithelium for genetic diagnosis of atherosclerosis using mtDNA mutations. Vessel Plus 2017;1:145-50.
73. Sazonova MA, Ryzhkova AI, Sinyov VV, et al. Mitochondrial mutations associated with cardiac angina. Vessel Plus 2019;3:8.
74. Sazonova MA, Sinyov VV, Ryzhkova AI, Sazonova MD, Khasanova ZB, Sobenin IA. MtDNA mutations linked with left ventricular hypertrophy. Vessel Plus 2019;3:5.
75. Sazonova MA, Ryzhkova AI, Sinyov VV, et al. New markers of atherosclerosis: a threshold level of heteroplasmy in mtDNA mutations. Vessel Plus 2017;1:182-91.
76. Sazonova MA, Sinyov VV, Barinova VA, et al. Mosaicism of mitochondrial genetic variation in atherosclerotic lesions of the human aorta. Biomed Res Int 2015;2015:825468.
77. Sobenin IA, Sazonova MA, Ivanova MM, et al. Mutation C3256T of mitochondrial genome in white blood cells: novel genetic marker of atherosclerosis and coronary heart disease. PLoS One 2012;7:e46573.
78. Sazonova MA, Zhelankin AV, Barinova VA, et al. Dataset of mitochondrial genome variants associated with asymptomatic atherosclerosis. Data Brief 2016;7:1570-5.
79. Orekhov AN, Poznyak AV, Sobenin IA, Nikifirov NN, Ivanova EA. Mitochondrion as a selective target for the treatment of atherosclerosis: role of mitochondrial DNA mutations and defective mitophagy in the pathogenesis of atherosclerosis and chronic inflammation. Curr Neuropharmacol 2020;18:1064-75.
80. Gammage PA, Viscomi C, Simard ML, et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat Med 2018;24:1691-5.
81. Gammage PA, Van Haute L, Minczuk M. Engineered mtZFNs for manipulation of human mitochondrial DNA heteroplasmy. Methods Mol Biol 2016;1351:145-62.
82. Yang Y, Wu H, Kang X, et al. Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein Cell 2018;9:283-97.
83. Gammage PA, Gaude E, Van Haute L, et al. Near-complete elimination of mutant mtDNA by iterative or dynamic dose-controlled treatment with mtZFNs. Nucleic Acids Res 2016;44:7804-16.
84. Bacman SR, Kauppila JHK, Pereira CV, et al. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat Med 2018;24:1696-700.
85. Hashimoto M, Bacman SR, Peralta S, et al. MitoTALEN: a general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol Ther 2015;23:1592-9.
86. Yahata N, Matsumoto Y, Omi M, Yamamoto N, Hata R. TALEN-mediated shift of mitochondrial DNA heteroplasmy in MELAS-iPSCs with m.13513G>A mutation. Sci Rep 2017;7:15557.
87. Pereira CV, Bacman SR, Arguello T, et al. mitoTev-TALE: a monomeric DNA editing enzyme to reduce mutant mitochondrial DNA levels. EMBO Mol Med 2018:10.
88. Zekonyte U, Bacman SR, Smith J, et al. Mitochondrial targeted meganuclease as a platform to eliminate mutant mtDNA in vivo. Nat Commun 2021;12:3210.
89. Sazonova MA, Ryzhkova AI, Sinyov VV, et al. Creation of cultures containing mutations linked with cardiovascular diseases using transfection and genome editing. Curr Pharm Des 2019;25:693-9.
90. Hussain SRA, Yalvac ME, Khoo B, Eckardt S, McLaughlin KJ. Adapting CRISPR/Cas9 system for targeting mitochondrial genome. Front Genet 2021;12:627050.
91. Loutre R, Heckel AM, Smirnova A, Entelis N, Tarassov I. Can mitochondrial DNA be CRISPRized: pro and contra. IUBMB Life 2018;70:1233-9.
92. Wang H, La Russa M, Qi LS. CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem 2016;85:227-64.
93. Antón Z, Mullally G, Ford HC, van der Kamp MW, Szczelkun MD, Lane JD. Mitochondrial import, health and mtDNA copy number variability seen when using type II and type V CRISPR effectors. J Cell Sci 2020;133:jcs248468.
94. Comte C, Tonin Y, Heckel-Mager AM, et al. Mitochondrial targeting of recombinant RNAs modulates the level of a heteroplasmic mutation in human mitochondrial DNA associated with Kearns Sayre Syndrome. Nucleic Acids Res 2013;41:418-33.
95. Sazonova MA, Sinyov VV, Ryzhkova AI, et al. Cybrid models of pathological cell processes in different diseases. Oxid Med Cell Longev 2018;2018:4647214.
96. Poursaleh A, Esfandiari G, Beigee FS, Eshghifar N, Najafi M. Isolation of intimal endothelial cells from the human thoracic aorta: study protocol. Med J Islam Repub Iran 2019;33:51.
97. Patel JJ, Srivastava S, Siow RCM. Isolation, culture, and characterization of vascular smooth muscle cells. In: Methods in molecular biology. Humana Press Inc. 2016; pp. 91-105.
98. Orekhov AN, Bobryshev YV. Cell composition of the subendothelial aortic intima and the role of alpha-smooth muscle actin expressing pericyte-like cells and smooth muscle cells in the development of atherosclerosis. In: Muscle cell and tissue. IntechOpen; 2015.
99. Lee H, Lee S, Baek G, et al. Mitochondrial DNA editing in mice with DddA-TALE fusion deaminases. Nat Commun 2021;12:1190.
100. Guo J, Zhang X, Chen X, et al. Precision modeling of mitochondrial diseases in zebrafish via DdCBE-mediated mtDNA base editing. Cell Discov 2021;7:78.
101. Gammage PA, Moraes CT, Minczuk M. Mitochondrial genome engineering: the revolution may not Be CRISPR-Ized. Trends Genet 2018;34:101-10.
102. Shepherd DL, Hathaway QA, Pinti MV, et al. Exploring the mitochondrial microRNA import pathway through Polynucleotide Phosphorylase (PNPase). J Mol Cell Cardiol 2017;110:15-25.
103. Chacinska A, van der Laan M, Mehnert CS, et al. Distinct forms of mitochondrial TOM-TIM supercomplexes define signal-dependent states of preprotein sorting. Mol Cell Biol 2010;30:307-18.
104. Bacman SR, Gammage PA, Minczuk M, et al. Manipulation of mitochondrial genes and mtDNA heteroplasmy. In: Pon LA, Schon EA, editors. Methods in cell biology. Mitochondria, 3rd Ed. Academic Press; 2020. pp. 441-87.
105. Zakirov FH, Zhang D, Grechko AV, Wu WK, Poznyak AV, Orekhov AN. Lipid-based gene delivery to macrophage mitochondria for atherosclerosis therapy. Pharmacol Res Perspect 2020;8:e00584.
106. Katayama T, Kinugawa S, Takada S, et al. A mitochondrial delivery system using liposome-based nanocarriers that target myoblast cells. Mitochondrion 2019;49:66-72.
107. Yamada Y, Akita H, Kamiya H, et al. MITO-Porter: A liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim Biophys Acta 2008;1778:423-32.
108. Ishikawa T, Somiya K, Munechika R, Harashima H, Yamada Y. Mitochondrial transgene expression via an artificial mitochondrial DNA vector in cells from a patient with a mitochondrial disease. J Control Release 2018;274:109-17.
109. Sinha S, Villarreal D, Shim EY, Lee SE. Risky business: Microhomology-mediated end joining. Mutat Res 2016;788:17-24.
110. Murugan K, Seetharam AS, Severin AJ, Sashital DG. CRISPR-Cas12a has widespread off-target and dsDNA-nicking effects. J Biol Chem 2020;295:5538-53.
111. Mok BY, Kotrys AV, Raguram A, Huang TP, Mootha VK, Liu DR. CRISPR-free base editors with enhanced activity and expanded targeting scope in mitochondrial and nuclear DNA. Nat Biotechnol 2022;40:1378-87.
112. Xiang G, Zhang X, An C, Cheng C, Wang H. Temperature effect on CRISPR-Cas9 mediated genome editing. J Genet Genomics 2017;44:199-205.
113. Chrétien D, Bénit P, Ha HH, et al. Mitochondria are physiologically maintained at close to 50 °C. PLoS Biol 2018;16:e2003992.
114. Mougiakos I, Mohanraju P, Bosma EF, et al. Characterizing a thermostable Cas9 for bacterial genome editing and silencing. Nat Commun 2017;8:1647.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Khotina VA, Bagheri Ekta M, Baig MS, Wu WK, Grechko AV, Sukhorukov VN. Challenges of mitochondrial DNA editing in mammalian cells: focus on treatment of cardiovascular disease. Vessel Plus 2022;6:65. http://dx.doi.org/10.20517/2574-1209.2022.28
AMA Style
Khotina VA, Bagheri Ekta M, Baig MS, Wu WK, Grechko AV, Sukhorukov VN. Challenges of mitochondrial DNA editing in mammalian cells: focus on treatment of cardiovascular disease. Vessel Plus. 2022; 6: 65. http://dx.doi.org/10.20517/2574-1209.2022.28
Chicago/Turabian Style
Khotina, Victoria A., Mariam Bagheri Ekta, Mirza S. Baig, Wei-Kai Wu, Andrey V. Grechko, Vasily N. Sukhorukov. 2022. "Challenges of mitochondrial DNA editing in mammalian cells: focus on treatment of cardiovascular disease" Vessel Plus. 6: 65. http://dx.doi.org/10.20517/2574-1209.2022.28
ACS Style
Khotina, VA.; Bagheri Ekta M.; Baig MS.; Wu W.K.; Grechko AV.; Sukhorukov VN. Challenges of mitochondrial DNA editing in mammalian cells: focus on treatment of cardiovascular disease. Vessel Plus. 2022, 6, 65. http://dx.doi.org/10.20517/2574-1209.2022.28
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 15 clicks
Cite This Article 15 clicks

 Like This Article 22
likes
Like This Article 22
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.