
Pharmacological agents affecting mitophagy and inflammation
 0
0 , ...
, ... Abstract
Mitochondria are cellular organelles providing energy to the cells. Due to the nature of mitochondrial enzymatic repair systems, mitochondrial DNA tends to generate mutations that are repaired less efficiently than nuclear DNA mutations. There is a certain relationship between the accumulation of mitochondria with mutated DNA in tissues, the development of oxidative stress, and several pathological conditions, from specific mitochondrial diseases to an increased risk of cancer, atherosclerosis, neurodegeneration, and non-systemic inflammation. Mitophagy is the biological mechanism responsible for the degradation of dysfunctional, damaged, and mutant mitochondria. Presumably, the stimulation of mitophagy can lead to tissue cleansing from dysfunctional mitochondria, which can have a powerful therapeutic effect on the root cause of the pathology. This review examines the relationship between mitochondrial mutations and the development of oxidative stress, the mechanisms of mitophagy, and a group of chemicals that stimulate mitophagy.
Keywords
INTRODUCTION
Each human cell contains from a few hundred to a couple of thousand independently multiplying mitochondria, united in a network of constantly merging and dividing organelles. Each mitochondrion contains 2-10 copies of the circular double-stranded mtDNA. It consists of 16,569 base pairs and encodes 37 genes, including 22 mitochondrial tRNA genes and 2 ribosomal RNA genes, which are required for the specific translation of 13 subunits of respiratory chain enzymes that are also encoded by mtDNA[1].
Due to a large number of redox reactions and spontaneous hydrolytic processes occurring in the mitochondria, as well as the constant production of ROS (reactive oxygen species) by the components of the respiratory chain and the abundance of nucleases, mtDNA is often damaged with the appearance of mutations in its structure[2].
Point nucleotide substitutions in the mitochondrial genome tend to occur due to improperly incorporated bases into the new circuit during mtDNA replication[3,4]. Most of the deletions in mtDNA appear in the so-called replication forks, which links their appearance with disruptions in processes of replication, reparation, and recombination[5]. It is assumed that, due to the absence of histones or similar proteins and the lower efficiency of enzymes involved in DNA reparation, the mitochondrial genome is much more vulnerable to the accumulation of mutations compared to the nuclear one[6].
Among all the congenital pathologies associated with metabolic disorders, mitochondrial diseases are the most common. Their frequency of occurrence is estimated at 1.6 per 5000[7]. In mitochondrial diseases, many tissues and organs are affected, and the most energy-demanding ones suffer the most, namely the central and peripheral nervous system, skeletal muscles, and the heart[7,8]. However, mitochondrial dysfunctions are associated with many other diseases, from liver diseases[9,10,11] to polycystic ovary syndrome[12], diabetes[13], and atherosclerosis[14].
Typically, these mutations are found in mtDNA, but beyond that, mitochondria-related pathologies can be caused by mutations in nuclear DNA, namely in genes encoding the approximately 1000 different mitochondrial proteins synthesized in the cytoplasm and transported to mitochondria[15].
Mitochondrial diseases are associated with a disruption of energy processes in the mitochondria. These violations also cause certain structural changes in these organoids. Among other things, in many cases, there is a connection between mitochondrial dysfunction and the development of inflammation or other pathological processes in tissues and organs, for example, atherosclerosis, although it is still unknown what the root cause is in this case[16].
Thus, mitochondrial quality control is important to prevent the development of chronic inflammation. The removal of entire mitochondria due to their dysfunction or aging is accomplished through a selective form of autophagy called mitophagy[17,18].
Mitophagy is a process aimed at the selective removal of damaged or excessive mitochondria, which ultimately allows cells to regulate their quantity and control their quality. Thus, mitochondria can be considered as morphologically dynamic structures undergoing continuous processes of fission and fusion, as well as degradation and mitochondrial biogenesis. Mitophagy is a key regulatory mechanism that limits cell damage due to the accumulation of mutated mtDNA and maintains tissue homeostasis[19,20]. Additionally, mitophagy is a special kind of macroautophagy, a conservative intracellular degradation mechanism that removes excess or dysfunctional cytoplasmic components and intracellular pathogens[21].
Since the accumulation of defective mitochondria leads to the damage of cells and tissues, they need to be disposed of. The process of mitophagy is evolutionarily conserved and is found in a wide variety of organisms, from yeast to mammals[22].
It has been shown that the decrease in mitochondrial function with age is associated with an alteration of the mitophagy process[23,24]. Thus, impaired mitophagy can be associated with a variety of pathological conditions, such as cardiovascular[25] and neurodegenerative diseases[26], myopathies[27], metabolic disorders[28], chronic inflammation[29], and oncology[30].
It can be assumed that the therapeutic effect aimed at increasing the level of mitophagy above the basal levels will be a promising direction in the treatment of various pathologies. At the moment, several mechanisms of mitophagy have been described as working through different signaling cascades. Mitophagy regulation can be ubiquitin-dependent or ubiquitin-independent. In this review, we discuss mechanisms of mitophagy and some agents modulating its activity.
MAIN TEXT
Mitochondrial damage leads to inflammation
An example of the interrelation between mitochondria and the development of inflammation in the nervous system is Leber hereditary optic neuropathy (LHON), a primary mitochondrial disease characterized by bilateral vision loss in early adulthood[31]. In 90%-95% of cases, this pathology develops due to mutations G3460A, G11778A, and T14484C, affecting the genes encoding subunits of the complex of the respiratory chain I. Dysfunction of complex I caused by these mutations in mtDNA leads to a decrease in ATP synthesis, an increase in ROS production, and disruption of glutamate transport, which leads to damage in the retina of the eye and dysfunctions of ganglion cells and their apoptosis[32]. However, patients may also experience other pathologies, in particular, damage to the peripheral optic nerves due to the development of an inflammatory process in them. However, it should be noted that mitochondrial dysfunction and the mutations that cause it can be either the cause of the development of neuroinflammation or the result of the nerve cell degeneration caused by it[31,32].
Another common mitochondrial disorder is MELAS syndrome (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes). Overall, 80% of patients diagnosed with this pathology are carriers of the mitochondrial mutation A3243G in the MT-TL1 gene, which encodes the mitochondrial tRNA of leucine and plays a key role in the translation of proteins needed for the proper assembly of mitochondrial complexes as part of the electron transport chain. Defects caused by this mutation damage the redox function of the electron transport chain, which leads to a lack of energy and oxidative stress, especially in cells with high energy needs, such as neurons and myocytes[33,34].
In addition, with MELAS, foci of inflammation are often observed in the tissues, particularly in the endothelium of large arteries, which indicates the relationship between mitochondrial dysfunction and the development of the inflammatory process, accompanied by oxidative stress. It is in such places that atherosclerotic lesions begin to form[34,35]. It is believed that the increased atherogenic risk detected in MELAS is primarily caused by increased ROS production[36] due to a disrupted electron transport chain.
The exact mechanism of the effect of mitochondrial mutations on vascular health is still unknown
The accumulation of mtDNA mutations can be accompanied by changes in mitochondrial structure and function, which can be observed under a microscope. Such altered mitochondria have been shown to be present in atherosclerotic lesions, suggesting a relationship between mtDNA mutations and the development of atherosclerosis with its further progression[37]. Moreover, it is the distribution of endotheliocytes with dysfunctional mitochondria in the endothelium of blood vessels that can at least partly explain the focal appearance of atherosclerotic lesions[16].
It should also be noted that an association of mutations of mitochondrial DNA with atherosclerotic lesions was found relatively recently[38,39], leading to the conclusion that there is a new potential mechanism of atherogenesis involving inflammation induced by mitochondrial DNA mutations[40,41]. Of course, the influence of multiple modified LDL, including oxidized and desialylated LDL[42,43,44] and their accumulation in arterial walls[45], on atherosclerosis pathogenicity should be taken into account[46]. In addition, components involved in the metabolism of LDL (e.g., LDL receptor and proprotein convertase subtilisin/kexin 9 (PCSK9), both involved in the process of removal of LDL from circulation) should also be taken into account. Interestingly, PCSK9, in addition to its role in cardiovascular disease[47], has a connection with the damage of mtDNA in the case of pro-inflammatory stimulation[48], and it is even related to certain types of cancer[49].
Another study showed that the content of the mitochondrial mutation G14459A is higher specifically in atherosclerotic lesions of human aortic intima, namely in fatty streaks and lipofibrous and fibrous plaques, compared to unaffected sites. This pattern is also confirmed for four other mutations (C3256T, G12315A, G13513A, and G15059A) that are also closely associated with cardiovascular disease risk[50]. Finally, an accumulation of deletions and single-nucleotide polymorphisms in mtDNA of patients with coronary heart disease compared with the control group has been shown[51]. The latter observation may be explained by the strong positive correlation between the presence of atherosclerosis and the development of coronary heart disease[52,53].
Such studies reveal only the relationship between the presence of a mutation load in the mitochondrial genome and the presence of cardiovascular disease, as well as the severity of its course. It is not even clear whether the described mtDNA mutations increase the predisposition to the development of atherosclerosis[37].
There are several hypotheses that explain such regularities. According to one of them, a decrease in the amount of normally functioning enzymes due to mutations in the genes encoding them in mitochondria may lead to oxidative damage to human vascular intima cells. The second one states that an increased mutation load of mtDNA can potentially cause disruption of cellular function and lead to the formation of oxidative stress conditions at the local level, which can cause the accumulation of lipoproteins[54,55] and simultaneously attract circulating monocytes and recruit them to the affected area[56,57,58], thus initiating the development of the pathological condition.
The mentioned association among MELAS, chronic inflammation, and risk of atherosclerosis was confirmed by experiments on endothelial cells derived from induced pluripotent stem cells (iPSCs) taken from a patient with MELAS with a high proportion of mitochondrial mutation A3243G. Their treatment with antioxidants such as vitamin C and edaravone successfully reduced the expression of inflammation markers, ASC (apoptosis-associated speck-like protein containing a CARD), an inflammasome adapter, and caspase-1, a proteolytic enzyme that activates pro-inflammatory cytokines by cleaving their precursors[35]. A decrease in monocyte adhesion on these cells has also been shown. Both inflammation[59] and the accumulation of phagocytes in the intima of large arteries with their subsequent activation[60] contribute to a pro-atherogenic phenotype[61]. In addition, impaired blood flow induced by endothelial dysfunction[62], which in turn can be caused by, for instance, excess production of ROS, is observed both in the early stages of atherosclerosis[63] and in other cardiovascular diseases.
Mitophagy functions as a mitochondrial quality control mechanism that allows selective destruction of damaged or unnecessary mitochondria[64,65]. This allows us to speak of this process as an effective means of protecting cells and tissues of the body from the reproduction of mutant mitochondria with impaired energy metabolism. Below, the main mechanisms of mitophagy, as well as the types observed under different conditions, are considered.
Mitophagy mediated by PINK1-Parkin
The most studied mechanism of mitophagy activation is the PINK1-Parkin pathway [Figure 1]. In properly functioning mitochondria, PINK1 kinase (PTEN-induced kinase 1) is transported across the mitochondrial inner membrane, where it is cleaved by several proteases. However, when the membrane potential is dissipated due to the damage of mitochondria, the inner membrane becomes depolarized and PINK1 begins to accumulate on the outer membrane[66]. At the same time, PINK1 is activated by autophosphorylation and stimulates the translocation of the cytoplasmic Parkin ligase to the mitochondrial surface[67]. In turn, Parkin ubiquitinates mitochondrial outer membrane proteins, such as MFN1, MFN2, and mitoNEET[68,69]. Ubiquitination of the mitochondrial surface is exactly the main factor signaling to the mitophagy adapter proteins and triggering the process of organoid uptake. It is unknown which proteins are necessary and sufficient for the initiation of mitophagy[22,70]. Mitophagy can be evaluated by reducing mitochondrial proteins or mRNA using immunoblotting, immunofluorescence, microscopy, or quantitative polymerase chain reaction[70,71]. In addition, the marker of mitophagy can serve as evidence of PINK1, Parkin, and their mRNA accumulation in the cell.
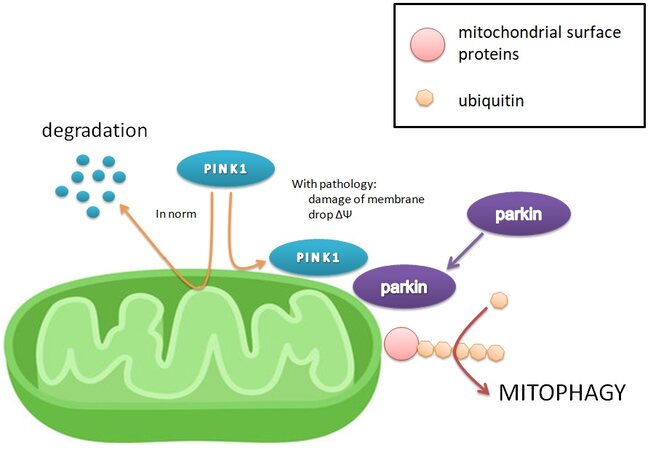
Figure 1. The mechanism of the main and most studied pathway of mitophagy activation, PINK1-Parkin. The drop in membrane potential leads to the accumulation of PINK1 on the outer mitochondrial membrane, whereas PINK1 is normally transported to the inner, where it degrades under the action of proteinases. The presence of PINK1 on the outer membrane indicates decreased membrane potential and damage to the mitochondria, and it attracts Parkin, which adds ubiquitin chains to surface mitochondrial proteins, serving as a signal to absorb mitochondria[22,66-71].
Parkin-independent mitophagy
Mitophagy is not always dependent on Parkin[65]. Other ligases of ubiquitin E3, such as Gp78, SMURF1, SIAH1, MUL1, and ARIH1, which are involved in the activation of mitophagy, are described[72,73]. They similarly ubiquitinate the surface of mitochondria by activating autophagy signaling molecules, including optineurin (OPTN), nuclear dot protein 52 kDa (NDP52), and p62[74]. These mitophagy induction pathways operate in parallel or in addition to the PINK1-Parkin pathway. Thanks to them, mitophagy is not disturbed by dysfunction or overexpression of PINK1 in cells. It is not yet known how the various pathways of mitophagy are regulated and interact with each other, as well as what contribution they make to maintaining the stability of the cell’s mitochondria[22,73].
In addition, there are specific mitophagy receptors on the surface of mitochondria, including Atg32 (Autophagy-related protein 32) in yeast and FKBP8 (FK506-binding protein 8), NIX (NIP3-like protein X), BNIP3 (BCL2 interacting protein 3), and FUNDC1 (FUN14 domain containing 1) in mammals.
There are also many ubiquitin-independent mitophagy receptors that reside on the outer mitochondrial membrane and stimulate mitochondrial uptake through direct interaction with LC3/GABARAP proteins[75,76].
FUNDC1 activity is inhibited by CK2 and Src kinases, which phosphorylate its LIR motif. When there is a need to start mitophagy, PGAM5 phosphatase dephosphorylates FUNDC1 to facilitate its interaction with LC3, while ULK1, on the contrary, phosphorylates it to enhance interaction with receptors on the phagosome surface. NIX and BNIP3 are activated by HIF1α and accumulate on the mitochondrial surface, thereby mediating mitophagy (more on this below in the section on iron chelators). In yeast, the only mitophagy receptor found, ATG32 is phosphorylated by CK2 to promote ATG11 interaction, leading to an association with ATG8 and mitochondrial recruitment to the phagosome assembly site[76].
The process of ubiquitin-independent mitophagy can be modulated at two levels, transcriptional and posttranslational. Thus, the regulation of the level of the receptor, for example, BNIP3 and NIX, is controlled by upstream mitophagy signals. Posttranslational modifications, primarily phosphorylation of certain residues, facilitate or increase the ability of the receptors to interact with LC3/GABARAP (although, in the case of FUNDC1, conversely, phosphorylation inhibits its activity under basic conditions). Other uncharacteristic posttranslational modifications may also be important in fine-tuning receptor activity[76].
Another mechanism that initiates mitophagy associated with a mitochondrion is cardiolipin, a specific lipid. In normally functioning mitochondria, cardiolipin is located in the inner membrane, where it interacts with proteins and participates in the maintenance of the structure of crista and stabilization of complexes of the respiratory chain. When the integrity of the mitochondria is damaged, cardiolipin translocates to the cytosolic side of the outer mitochondrial membrane and binds to the autophagosome receptor LC-II, which leads to mitophagy by mitochondrial uptake[77,78,79].
It has been shown that G2019S mutation in PARK8 gene coding for LRRK2 (leucine-rich repeat kinase 2), leading to the repression of basal mitophagy and associated with Parkinson’s disease, can be suppressed with the help of LRRK2 kinase inhibitor[80].
The PINK1-Parkin pathway may not be the main mitophagy pathway in vivo
Although the PINK1-Parkin pathway is considered canonical, it is turned on when the mitochondrial membrane potential is dissipated and is a response to severe damage to these organelles. The literature mentions many ways to stimulate it under artificial conditions, but it does not seem to be the main way of mitophagy in vivo, both in mice and Drosophila.
Until recently, the PINK1-Parkin pathway has been studied in cell cultures, so its function in vivo is still largely unclear. However, experiments conducted on animal models of mice and fruit flies with a knockout of the genes participating in the PINK1-Parkin pathway showed that, in the presence of various disorders in the development of nervous and muscle tissues, as well as with a certain deficit in motor activity, the base mitophagy level in their tissues remains virtually unchanged[81,82,83].
These results may indicate that the PINK1-Parkin pathway does not actually play an important role in maintaining a baseline level of mitophagy, which may cast doubt on the relevance of a pharmacological intervention on this pathway. Nevertheless, it is known that, in many pathological conditions associated specifically with a decrease in mitophagy, this particular pathway is damaged[84-87], which indicates the importance of this signaling pathway for the maintenance of normal functioning of mitochondria and cells in general. Thus, we believe that this method of influencing mitophagy is still relevant.
Types of mitophagy
Depending on the physiological context, basal, stress-induced, and programmed mitophagy are distinguished.
Basal mitophagy is always present in cells, ensuring the utilization of old or damaged organelles. However, its degree varies between tissues and even among different types of cells in the same tissue, which suggests cellular autonomic regulation. In some cases, basal mitophagy is independent of PINK1, and it appears that the factors controlling it are tissue specific[24,81,88].
Stress-induced mitophagy contributes to the quality control of mitochondria in extreme conditions for the cell, as well as mediates metabolic adaptation to external influences. Thus, the dissectors of the mitochondrial respiratory chain induce mitophagy by activating the PINK1-Parkin pathway[89]. Nitrogen or iron deficiency, as well as hypoxia, causes mitophagy through the activation of specific receptors located on the surface of mitochondria[90,91].
Programmed mitophagy is characteristic of several cells that must eliminate their mitochondria during development. For example, erythrocytes remove their organelles during maturation[92,93]. Fertilized oocytes in nematodes, flies, and mice need to lyse sperm-derived mitochondria[94,95,96]. Under natural hypoxic conditions, fetal cardiomyocytes are forced to switch to glycolysis and destroy some of their mitochondria by activating the PINK1-Parkin pathway for normal development and functioning[97]. During the development of the retina, mitophagy is activated in ganglion cells to switch towards glycolysis, which, in turn, contributes to the development of cell differentiation[98]. At the same time, they activate a separate mitophagy pathway associated with the activation of HIF1/BNIP3L/NIX under hypoxic conditions. During the polarization of macrophages towards the pro-inflammatory M1 phenotype, predominantly using glycolysis, NIX-dependent mitophagy is activated, which promotes a high elimination of mitochondria. When macrophages are polarized along the M2 pathway, the functioning of which depends on oxidative phosphorylation, NIX-dependent mitophagy does not occur[98].
Significance of mitophagy
Impaired mitophagy is associated with aging and many pathological conditions, such as cardiovascular and neurodegenerative diseases, myopathies, metabolic disorders, inflammation, and cancer.
It has been shown that impaired functioning of mitochondria leads to hyperproduction of ROS; oxidation of lipids, nucleic acids, and proteins; and activation of the NLRP3 inflammasome, which in turn can aggravate chronic inflammation as well as cause the development of atherosclerosis through endothelial damage and stimulation of increased accumulation of cholesterol in macrophages. This is induced by atherogenic multiple-modified LDL (low density lipoprotein) in the artery wall and eventually leads to the formation of atherosclerotic lesions and their maturation[99,100].
It can be assumed that substances that enhance mitophagy in tissues relative to the baseline level can become the basis for the treatment of a wide range of pathologies associated with inflammation as well as for the ones directly related to the presence of mitochondrial DNA mutations. At the moment, many substances have been discovered to affect the processes of mitophagy. According to the mechanism of action, they can be divided into several groups, which are described below.
Protonophores and mitochondrial toxins
Protonophores are weakly acidic lipophilic compounds that can transport protons through the inner mitochondrial membrane, which leads to a drop in the gradient of the electrochemical potential of the mitochondria and the disconnection of oxidative phosphorylation with the electron transport chain. As a result of significant leakage of protons, mitochondria undergo mitophagy, mainly through activation of the PINK1-Parkin pathway[101].
Despite their widespread use in basic research, protonophores are poorly suited for the role of chemical stimulators of mitophagy. In particular, the effects they have on mitochondria are not similar to those that occur naturally, are largely detrimental, and ultimately lead to mitochondrial insufficiency. In addition, they affect the entire population of mitochondria, rather than a specific subgroup (e.g., those with mitochondrial mutations), are non-specific and have protonophoric activity on other membranes[101,102].
An example of a protonophore is the substance FCCP (trifluoromethoxy carbonylcyanide phenylhydrazone), otherwise called CCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone). On a model of HeLa cells consistently expressing YFP-Parkin, FCCP has been shown to increase the permeability of the internal mitochondrial membrane to H+, which leads to its depolarization and an increase in the level of PINK1 in the mitochondrial fraction in the absence of changes in the cytosol fraction. The accumulation of PINK1 begins 30 min after the treatment of cells with FCCP and continues for 3 h[103].
Another example of a protonophore is 2,4-dinitrophenol (DNP), which was once used as a remedy for obesity but was withdrawn from clinical practice due to high toxicity and side effects.
DNP affects the activity of the electron transport chain and causes the release of protons into the mitochondrial matrix [Figure 2]. This substance also converts the cell to an alternative production of ATP and, among other things, reduces the formation of ROS, which damages organelles, as well as promotes mitochondrial biogenesis[104,105].
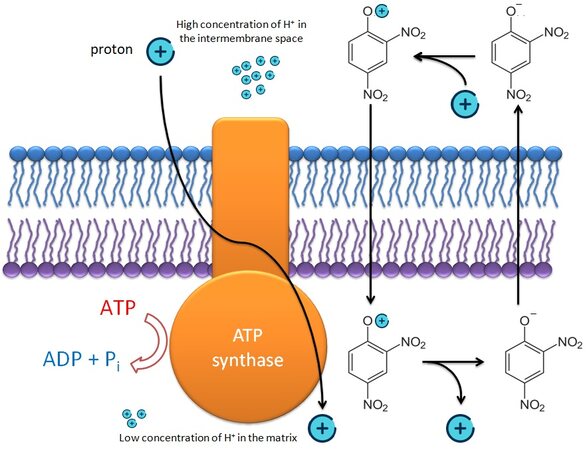
Figure 2. 2,4-dinitrophenol acts as an uncoupler of oxidative phosphorylation. Due to its hydrophobic properties, it freely penetrates through the inner mitochondrial membrane, transporting protons along the concentration gradient bypassing ATP synthase, which leads to a drop in membrane potential and a decrease in energy production in the cell[104,105].
It has been shown that the injection of DNP stimulates several signaling pathways for the stress response in neurons. In cultures of cortical neurons and animal models, it has been demonstrated that low doses of DNP and the resultant mild mitochondrial uncoupling can protect neurons from oxidative stress, excitotoxicity, and accumulation of neurodegenerative disease-related self-aggregation proteins, such as amyloid b-peptide, tau, and a-synuclein. Thus, DNP is considered a promising treatment for reducing brain damage in Alzheimer’s disease, Parkinson’s disease, epilepsy, and cerebral ischemic stroke[105].
The so-called second-generation protonophores have less cytotoxicity and do not depolarize the plasma membrane, but their effect on mitophagy processes has not yet been studied. Such substances include the recently synthesized BAM15, which causes a stable soft uncoupling of mitochondrial membranes. This substance is less cytotoxic compared to FCCP and DNP[106].
In addition, adding BAM15 to the culture of macrophages has been shown to reduce their production of pro-inflammatory cytokines and enhance anti-inflammatory polarization of the M2 type, which allows it to be considered as an effective means to control immune responses[107].
Mitochondrial toxins are a more general group of substances that inhibit respiratory processes and thereby activate PINK1-mediated mitophagy[101].
A typical example of such a substance is the cyclopeptide antibiotic valinomycin, which is a highly specific lipophilic ionophore capable of transporting K+ ions across the inner membrane[108], eliminating the electrical potential and activating the PINK1-Parkin pathway without changing the membrane potential
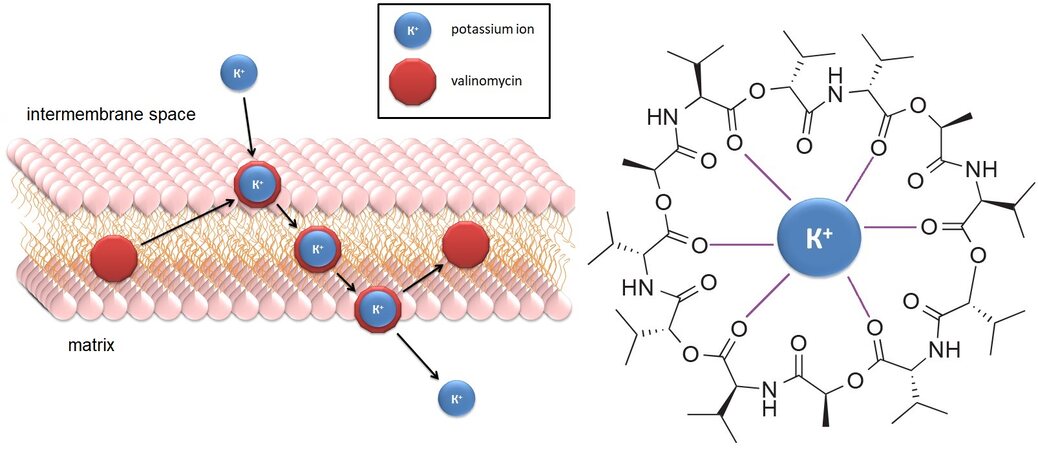
Salinomycin is a registered antibiotic, as well as a potential selective anti-cancer agent. It is thought to act as an ionophore for K+, but, unlike valinomycin, salinomycin causes rapid hyperpolarization by mediating K+/H+ exchange through the inner mitochondrial membrane. Thus, it causes the outflow of K+ ions from mitochondria and a decrease in their membrane potential[111].
Treatment of intact mouse embryonic fibroblasts (MEF) and HMLE cancer stem cells with salinomycin causes acidification of the mitochondrial matrix and a significant decrease in respiration (with no increase in the production of ROS) for tens of minutes. All these effects appear a few seconds after the drug is applied but are short-term and completely disappear 12-48 h after the addition of the drug[112].
Valinomycin and salinomycin carry other ions rather than protons through mitochondrial membranes. They are no longer so radical as FCCP but produce similar effects, including the stimulation of mitophagy, allowing their potential usage for medical purposes.
A similar pattern is observed when cells are treated with another potassium ionophore, nigericin. Both substances stimulate autophagy in the cells and, ultimately, cause apoptosis, which allows them to be considered promising drugs in oncology[112].
PINL1-cleaving protease inhibitors
The activity of the PINK1-Parkin pathway is also controlled by a number of proteases that catalytically cleave PINK1 and thus affect the mitophagy process[113]. Their inhibition by pharmacological agents may enhance mitophagy and be of clinical significance.
An example of such a protease is the transmembrane protease PARL (PINK1/PGAM5-associated rhomboid-like protease), which is located on the inner mitochondrial membrane of mammals and catalyzes the cleavage of PINK1, thereby controlling its content and localization in the cell[114,115,116]. In addition to regulating mitophagy, PARL is also involved in important cellular events such as apoptosis and regulation of mitochondrial dynamics[116].
PARL is a member of a family of diamond-shaped intramembrane proteases, which are membrane-embedded serine peptidases whose active sites are located in the bilayer of cell membranes[117,118].
There are currently no known selective inhibitors of mammalian rhomboid proteases, including PARL, that would be suitable for use in biological research, let alone therapeutic applications. However, it has recently been found that several succinimide-containing sulfonyl esters and sulfonamides are able to reversibly bind to PARL and inhibit its activity, with the most potent sulfonamides possessing submicromolar affinities for the enzyme[119]. These data may help to develop a group of new specific inhibitors of rhomboid proteases. In the future, they could be used not only for studying rhomboid proteases functions, which remain unknown for a number of enzymes of this family, but also for exploring their effect on mitophagy and evaluating the therapeutic effect.
Modulators of the PINK1-Parkin Pathway
The PINK1-Parkin signaling pathway plays a key role in removing damaged mitochondria. It is known that several mutant proteins affecting catalytic activity involved in this pathway can lead to the disruption of mitophagy processes and cause the development of one of the rare forms of Parkinson’s disease with early onset[101].
The structure of PINK1 contains regions whose homologs in other proteins provide contact with the adenine ring from ATP. This suggests that nucleotides other than ATP can bind to PINK1 and thereby alter its stability and function. The metabolic precursor of such a neo-substrate can be taken up by cells and converted into the form of nucleotide triphosphate, which would eventually lead to accelerated recruitment of Parkin to depolarized mitochondria[120,121].
An example of such a substance is kinetin, a plant hormone and derivative of adenine, which can be found in the tissues of all studied organisms, from plants to humans. Its ability to induce mitophagy was also confirmed in experiments with cardiac myoblast cells (H9c2). Treatment of cells by the active substance with the subsequent isolation of the mitochondrial fraction from them and analysis of the expression levels of MAP1LC3-II (Microtubule-associated proteins 1A/1B light chain 3A), a marker of autophagic vesicles, showed an acceleration of mitophagy processes under the influence of kinetin[122].
Iron chelators
It has been shown that the binding of iron ions, and, thereby, a decrease in their content in the cell, leads to the induction of mitophagy. At the same time, a transition from oxidative phosphorylation to glycolysis is observed in the cells, which indicates the cessation of respiratory processes[101].
The mechanism of mitophagy induced by iron chelation includes the stabilization of HIF1α[123], a subunit of the transcription factor HIF1 (hypoxia-inducible factor-1), which is hydroxylated and proteolyzed in the cytoplasm under normoxic conditions. However, under hypoxic cleavage conditions, this molecule ceases to be destroyed, transfers to the nucleus, and binds to the HIF1β subunit to activate its target genes[124].
In addition to being involved in response to hypoxic stress, HIF1α is also involved in the activation of other targets, namely the mitochondrial outer membrane anchor proteins, BNIP3 and NIX. These HIF1α-regulated molecules have a similar role in marking mitochondria for mitophagy, as does ubiquitylation of mitochondrial outer membrane proteins during depolarization-induced mitophagy, which explains the lack of requirement for PINK1 and Parkin in this mitophagy pathway[123]. It was found that HIF1α-dependent mitophagy is capable of facilitating differentiation of cardiomyoblasts via upregulation of BNIP3 and NIX[125].
Thus, HIF1α is the “major regulator” of iron depletion (and hypoxia)-induced mitophagy, controlling mitochondrial labeling through activation of BNIP3 and NIX while simultaneously regulating cellular metabolic activity.
Most iron chelators cause a drop in membrane potential, which leads to their toxicity for mitochondria and the cell as a whole. Thus, the iron ion chelator substance 1,10-phenanthroline (Phen) confirmed its effectiveness among 1200 chemical compounds applied to HeLa line cells to discover a new mitophagy modulator. Furthermore, it has been shown that the treatment of HeLa line cells with Phen increases the percentage of fragmented mitochondria after 4 h, increases the depolarization of mitochondrial membranes, and reduces ATP levels after 48 h. In this case, fragmentation of mitochondria and increased production of ROS along with suppression of cell division are observed[126].
However, there are exceptions; not all chelators cause such strong membrane uncoupling. For example, deferiprone (DFP) can bind iron ions, which leads to the renewal of mitochondria and the protection of cells from oxidative stress without affecting the membrane potential. At the same time, it retains the ability to promote mitophagy in cells with dysfunction of the PINK1-Parkin pathway[127,128].
Experiments with primary human fibroblasts, as well as with fibroblasts isolated from patients with Parkinson’s disease and containing Parkin mutations, showed that iron binding by chelators, for example, deferiprone, specifically induces mitophagy and not general autophagy. A correlation was observed between levels of mitophagy and transferrin receptors, which differed depending on the dose and even the type of substance introduced, which confirms the role of iron loss in the stimulation of mitophagy[123].
At the same time, treatment of cells with deferiprone does not lead to an increase in the content of PINK1, and the removal of PINK1 from cells using siRNA does not change the level of mitophagy when exposed to DFP[123]. In addition, iron chelation induces mitophagy in HeLa cells[104], which are reported to not express Parkin[129]. All this indicates that the PINK1-Parkin pathway is not activated by iron chelation and a different, Parkin-independent mitophagy is involved here.
It should be noted that the addition of deferiprone to cells leads to a profound restructuring of their metabolism, as well as causes DGAT1-dependent biosynthesis of lipid droplets [diacylglycerol acyltransferase (DGAT)], observed several hours before the destruction of mitochondria. Iron-induced lipid droplet biogenesis occurs independently of selective autophagy. It always precedes the removal of damaged mitochondria, and nascent lipid droplets tend to cluster within the mitochondrial network until PINK1-Parkin-independent elimination. At the same time, inhibition of DGAT1 limits mitophagy in vitro with disruption of lysosomal homeostasis and cell viability, which suggests an unexpected synergy between lipid homeostasis and mitophagy[130].
A possible reason that iron loss can lead to mitophagy may be the need for this element for the normal functioning of many mitochondrial enzymes, particularly all four respiratory chain complexes[131]. After all, it is the mitochondria that produce the iron-sulfur clusters and heme groups that the cell so greatly needs[132,133]. Further research into the relationship between iron levels and mitochondrial health may be promising both from the point of basic science and pharmacology that stimulates mitophagy.
AMPK-dependent path regulators
The enzyme AMPK (AMP-activated protein kinase) induces autophagy by direct phosphorylation of unc-51-like kinase (ULK1), a key initiator of the autophagic process. In turn, activated ULK1 interacts with the FUNDC1 protein localized in the mitochondrial membrane and thereby promotes the movement of mitochondria into autophagosomes[134].
Metformin is a glucose-lowering drug widely used to treat type II diabetes[135]. Among other things, it can induce mitophagy by affecting AMPK[136], which is accompanied by the formation of acidic vesicles and mitophagosomes and an increase in the level of mitophagy marker proteins: components of several pathways of mitophagy, such as PINK1, Parkin, and NIX, and specific membranes of lysosome proteins, e.g., LAMP2 and MAP1LC3B[137].
The study of the effect of metformin on phosphorylation and activation of AMPK was conducted on the primary culture of monocytes of healthy people. After exposure to metformin, there was a significant decrease in mitochondrial mass, an increase in the level of LC3-II (autophagosomal marker, form LC3-phosphatidylethanolamine conjugate, which is recruited to autophagosomal membranes) and several mRNAs of mitophagy proteins, a decrease in p62, and an increase in the formation of mitophagosomes[134].
SIRT1 Activators
SIRT1 (sirtuin 1) is an NAD+-dependent protein deacetylase that regulates transcription processes and plays a key role in controlling metabolism and differentiating and repairing DNA, as well as indirectly affecting inflammation. SIRT1 promotes cell survival by reducing oxidative stress and increasing mitochondrial division, which is achieved by affecting the transcription factors of the forkhead box O (FOXO) transcription factor and the coactivator of the peroxisome PPAR (proliferator-activated receptor gamma), which affects the activity of peroxisomes[138].
Nicotinamide (NAM) is known to promote mitophagy without impairing mitochondrial function by disruption of its membrane potential. It is a biosynthetic precursor of NAD+, which, in turn, activates NAD+-dependent sirt1 deacetylase (sirtuin 1, SIRT1) and thereby promotes the removal of damaged mitochondria from the cell[101].
The effect of NAM on mitophagy has been shown in human fibroblast culture. Its effect is conducted by means of an increase in the ratio of [NAD]/[NADH]. Cells cultured in an environment with the addition of 5 mM NAM every three or four days show a decrease in mitochondrial content in the first three days after the start of treatment, as detected by flow cytometry. In the following days, their level remains at 70% of the number of mitochondria in unprocessed cells (ordinary fibroblasts and mcF-7, H460, and HCT116 cancer cell lines)[139].
In addition, the ratio of [NAD]/[NADH] can be raised by adding asparagine (Asn) to the cells, which mobilizes the aspartate–malate shuttle in the direction of the increase of asparagine content. As a result, there is a decrease in the number of mitochondria. Moreover, excess NAM, NAD, and Asn induce fragmentation of mitochondria, which is a necessary prerequisite for mitophagy[139].
Curiously, a decrease in the ratio of [NAD]/[NADH], on the contrary, enhances the biogenesis of mitochondria and thereby contributes to their accumulation. This, in particular, occurs when lactate is added to cell culture[139].
Other SIRT1 activators, such as the natural substances resveratrol and fisetin, the synthetic small molecule SRT1720, also reduce the number of mitochondria in the cell without impairing their membrane potential. Perhaps mitophagy through SIRT1 can be carried out exclusively by the receptor mechanism and does not require the involvement of the PINK1-Parkin pathway[101].
Resveratrol (3,5,4’-trihydroxy-trans-stilbene) is a natural polyphenol synthesized by some plants as a protective reaction against parasites. It is found in large quantities in the peel and seeds of grapes, as well as red wine. It is an activator of SIRT1; under its influence, damaged mitochondria are destroyed in cells, resulting in a decrease in the level of ROS. Additionally, because of its hydrophobicity, it can accumulate in lipid formations, for example, in cell membranes[140,141].
The mitophagy effect of resveratrol was detected in experiments on an immortalized line of mouse myoblastic cells (C2C12). The addition of 30 μM resveratrol to the culture resulted in a decrease in mitochondrial mass. The effect is offset by the transfection of siRNA PINK1 cells (while the transfection of the control siRNA has no effect) or the addition of 50 μM chloroquine (SQ), which inhibits the synthesis of nucleic acids[140].
Fisetin is a flavonoid present in many plants as a yellow-ochre dye. It is also found in large quantities in vegetables and fruits such as strawberries, apples, persimmons, onions, and cucumbers[142.143]. As with many other polyphenols, it can activate SIRT1[143].
Fisetin can inhibit the proliferation of PANC-1 pancreatic cancer cells, one of the most aggressive and most resistant cell lines among pancreatic cancers. In this case, fisetin induces apoptosis and stimulates the formation of autophagosomes, which ultimately gives it a cytoprotective effect[143].
Treatment of cells with fisetin leads to an increase in the expression of marker proteins of mitophagy, namely PERK, ATF4, ATF6, PINK1, and Parkin, as well as to the accumulation of LC3-II on the mitochondria, as detected by immunomicroscopy. All these phenomena begin to appear as early as 12 h after the injection of fisetin[143].
Quercetin is a natural flavonoid with a powerful antioxidant effect. Among other things, it helps reduce mitochondrial oxidative stress, as well as enhance mitochondrial biogenesis[144,145].
The effect of quercetin on mitophagy has been demonstrated in the rat proximal tubule epithelial cell line NRK-52E. The cells, pretreated with quercetin (20 μM) and/or Ex-527 (1 μM), a SIRT1 inhibitor, were stimulated with angiotensin II (100 nM) for 12 h. In this case, joint localization of Parkin and PINK1 was observed, which confirms the activation of mitophagy by quercetin through its effect on the activity of SIRT[146].
Western blotting showed that quercetin treatment markedly enhanced the expression of mitophagy markers LC3-II, PINK1, and Parkin and reduced p62 expression levels compared to those in the control group, while EX-527 weakened this effect[146].
Caloric Restriction Mimetics
Calorie restriction (CR), that is, reducing calorie intake without malnutrition, can effectively counteract the features associated with aging, including a decrease in autophagy in cells. As a result, it prevents the accumulation of damaged mitochondria in cells of tissues. Due to the complexity of applying this approach to humans, several efforts are aimed at finding so-called caloric restriction mimetics (CRM) - pharmacological agents capable of reproducing the basic biochemical properties of CR, including reducing levels of protein acetylation and inducing autophagy in tissues[147].
Thus, the well-known drug aspirin and its active metabolite salicylate can induce autophagy due to their inhibitory effect on acetyltransferase EP300 (E1A-associated protein p300). This effect is achieved through competition between salicylate and acetyl coenzyme A for binding to the enzyme’s catalytic domain, and it has been observed in a wide variety of organisms, from the nematode Caenorhabditis elegans to mice[147].
The effectiveness of aspirin as an autophagy stimulant has been shown on human colorectal cancer cells (HCT116) and the nematode Caenorhabditis elegans. HCT116 cells were incubated for 16 h in the presence of increasing concentrations of salicylate. As a result, conversion of LC3-I to LC3-II was observed, indicating activation of autophagy and stimulation of mitophagy. A similar effect was observed in experiments with mice, in which the accumulation of LC3-II in the heart and liver was recorded 6 h after the administration of the aspirin dose[148].
Spermidine is a small molecule, aliphatic polyamine found in the ribosomes of a wide variety of organisms and performs various metabolic functions. Its effect on increasing mitophagy levels and lifespan has been shown in a variety of model organisms, such as mice, C. elegans, yeast, and human fibroblasts. The effect of spermidine on the inhibition of the activity of histone acetyltransferases, such as Iki3 p and Sas3p, causing hypoacetylation of histone H3, has been shown. A decrease in their activity can affect the epigenetic regulation of gene transcription, which allows the induction of transcripts relevant to autophagy[149]. In addition, it was found that, in C. elegans with turned-off genes PINK1 and PDR-1 by RNA interference, the effect of spermidine disappears, indicating a possible mechanism of action of this substance[149].
CONCLUSION
Disorders of mitophagy underlie many pathological processes, including the development of non-systemic inflammation and atherosclerosis. It is assumed that the development of these pathologies is associated with certain mitochondrial mutations, the appearance of which not only leads to metabolic disorders in the form of a drop in ATP levels and increased production of ROS but also reduces the effectiveness of mitophagy, preventing loss of mutant mitochondria.
We suggest the stimulation of mitophagy with a specially selected pharmacological agent capable of leading to a decrease in the number of mutant mitochondria, which in theory can contribute to the cleaning of tissues from damaged mitochondria and thereby lead to an improvement in the patient’s condition by removing the cause of the development of the pathological process. Thus, the study of substances that cause mitophagy and their mechanisms of action presents an important scientific task that can help in the search for drugs for a variety of diseases.
Today, many substances that are stimulants of mitophagy have been discovered. They differ not only in their chemical structures, but also in the targets of exposure to various processes occurring in the cell. Thus, mitochondrial uncouplers cause depolarization of mitochondrial membranes and, in turn, reduce the efficiency of energy metabolism. This entails the ubiquitination of the surfaces of mitochondria by Parkin or other ubiquitin ligases, which become a signal to mitophagy.
Proton transporters, such as FCCP and DNP, are highly toxic to cells, and, although their moderate doses have beneficial effects in cellular and even animal models, their use in clinical practice looks doubtful. However, at the moment, substances with a similar effect, causing the so-called mitochondrial uncoupling, which also leads to mitochondria elimination, have been synthesized and are not so dangerous for cells and the body as a whole. The influence of new generation protonophores specifically on mitophagy is still unknown, but they look quite promising in a new quality.
Other promising candidates are substances that act on signaling pathways that directly or indirectly affect mitophagy without depolarization of the mitochondrial membrane. Some of them activate the PINK1-Parkin pathway (kinetin and synthesized non-standard nucleotides, inhibitors of proteases, that cleave PINK1), one regulates and modulates the activity of the AMPK enzyme (metformin), and others increase the activity of the enzyme SIRT1 (nicotinamide, resveratrol, fisetin, and quercetin). Finally, when aspirin inhibits EP300 acetyltransferase, cells begin to digest their mitochondria, which can also be used in clinical practice. In addition, the small molecule spermidine also activates autophagy in tissues, and the lack of spermidine due to aging is considered one of the reasons for the accumulation of damaged mitochondria in tissues.
These substances are not toxic, largely due to the lack of effects on the processes of respiration and energy production in mitochondria. Some of them are natural for humans (kinetin, nicotinamide, and spermidine), others enter the organism with food (plant metabolites such as resveratrol and fisetin), and some have long been used in medicine and do not give side effects when used correctly (metformin and aspirin). It is possible that new data on these substances, as well as their effect on mitophagy, will allow us to develop approaches to the treatment of several pathologies caused by the accumulation of mutant mitochondria.
DECLARATIONS
Authors’ contributionsFirst author, concept and design, writing (original draft preparation): Borisov E
Writing (review & editing), supervision, critical review, project administration: Bezsonov E
Writing (review & editing), data source curating: Lyukmanov D
Writing (visualizationpreparation): Poggio P
Writing (visualization preparation): Moschetta D
Writing (visualization preparation): Valerio V
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe work was supported by the Russian Science Foundation, grant number 22-25-00480.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Authors 2022.
REFERENCES
1. der Bliek AM, Sedensky MM, Morgan PG. Cell biology of the mitochondrion. Genetics 2017;207:843-71.
2. Keogh MJ, Chinnery PF. Mitochondrial DNA mutations in neurodegeneration. Biochim Biophys Acta 2015;1847:1401-11.
3. Baines HL, Stewart JB, Stamp C, et al. Similar patterns of clonally expanded somatic mtDNA mutations in the colon of heterozygous mtDNA mutator mice and ageing humans. Mech Ageing Dev 2014;139:22-30.
4. Simsek D, Furda A, Gao Y, et al. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 2011;471:245-8.
5. Kazak L, Reyes A, Holt IJ. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 2012;13:659-71.
7. Tan J, Wagner M, Stenton SL, et al. Lifetime risk of autosomal recessive mitochondrial disorders calculated from genetic databases. EBioMedicine 2020;54:102730.
8. Frazier AE, Thorburn DR, Compton AG. Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology. J Biol Chem 2019;294:5386-95.
9. Dabravolski SA, Bezsonov EE, Baig MS, Popkova TV, Orekhov AN. Mitochondrial lipid homeostasis at the crossroads of liver and heart diseases. Int J Mol Sci 2021;22:6949.
10. Dabravolski SA, Bezsonov EE, Orekhov AN. The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed Pharmacother 2021;142:112041.
11. Dabravolski SA, Bezsonov EE, Baig MS, et al. Mitochondrial mutations and genetic factors determining NAFLD risk. Int J Mol Sci 2021;22:4459.
12. Dabravolski SA, Nikiforov NG, Eid AH, et al. Mitochondrial dysfunction and chronic inflammation in polycystic ovary syndrome. Int J Mol Sci 2021;22:3923.
13. Dabravolski SA, Orekhova VA, Baig MS, et al. The role of mitochondrial mutations and chronic inflammation in diabetes. Int J Mol Sci 2021;22:6733.
14. Salnikova D, Orekhova V, Grechko A, et al. Mitochondrial dysfunction in vascular wall cells and its role in atherosclerosis. Int J Mol Sci 2021;22:8990.
15. Stenton SL, Prokisch H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020;56:102784.
16. Sobenin IA, Sazonova MA, Postnov AY, Bobryshev YV, Orekhov AN. Changes of mitochondria in atherosclerosis: possible determinant in the pathogenesis of the disease. Atherosclerosis 2013;227:283-8.
17. Gottlieb RA, Thomas A. Mitophagy and mitochondrial quality control mechanisms in the heart. Curr Pathobiol Rep 2017;5:161-9.
18. Gkikas I, Palikaras K, Tavernarakis N. The role of mitophagy in innate immunity. Front Immunol 2018;9:1283.
19. Liesa M, Palacín M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 2009;89:799-845.
20. Mouli PK, Twig G, Shirihai OS. Frequency and selectivity of mitochondrial fusion are key to its quality maintenance function. Biophys J 2009;96:3509-18.
21. Chen G, Kroemer G, Kepp O. Mitophagy: an emerging role in aging and age-associated diseases. Front Cell Dev Biol 2020;8:200.
22. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 2018;20:1013-22.
23. Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015;521:525-8.
25. Pedro JM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res 2017;120:1812-24.
26. Mani S, Swargiary G, Chadha R. Mitophagy impairment in neurodegenerative diseases: Pathogenesis and therapeutic interventions. Mitochondrion 2021;57:270-93.
27. Borgia D, Malena A, Spinazzi M, et al. Increased mitophagy in the skeletal muscle of spinal and bulbar muscular atrophy patients. Hum Mol Genet 2017;26:1087-103.
28. Chen Z, Berquez M, Luciani A. Mitochondria, mitophagy, and metabolic disease: towards assembling the puzzle. Cell Stress 2020;4:147-50.
29. Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2020;16:3-17.
30. Denisenko TV, Gogvadze V, Zhivotovsky B. Mitophagy in carcinogenesis and cancer treatment. Discov Oncol 2021;12:58.
31. Yu-Wai-Man P, Turnbull DM, Chinnery PF. Leber hereditary optic neuropathy. J Med Genet 2002;39:162-9.
32. Bargiela D, Chinnery PF. Mitochondria in neuroinflammation - multiple sclerosis (MS), leber hereditary optic neuropathy (LHON) and LHON-MS. Neurosci Lett 2019;710:132932.
33. Murakami H, Ono K. MELAS: mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes. Brain Nerve 2017;69:111-7.
34. Seitun S, Massobrio L, Rubegni A, et al. MELAS syndrome with cardiac involvement: a multimodality imaging approach. Case Rep Cardiol 2016;2016:1490181.
35. Pek NMQ, Phua QH, Ho BX, et al. Mitochondrial 3243A > G mutation confers pro-atherogenic and pro-inflammatory properties in MELAS iPS derived endothelial cells. Cell Death Dis 2019;10:802.
36. Chistiakov DA, Revin VV, Sobenin IA, Orekhov AN, Bobryshev YV. Vascular endothelium: functioning in norm, changes in atherosclerosis and current dietary approaches to improve endothelial function. Mini Rev Med Chem 2015;15:338-50.
37. Poznyak AV, Ivanova EA, Sobenin IA, Yet SF, Orekhov AN. The role of mitochondria in cardiovascular diseases. Biology (Basel) 2020;9:137.
38. Sobenin IA, Sazonova MA, Postnov AY, Salonen JT, Bobryshev YV, Orekhov AN. Association of mitochondrial genetic variation with carotid atherosclerosis. PLoS One 2013;8:e68070.
39. Sobenin IA, Zhelankin AV, Khasanova ZB, et al. Heteroplasmic variants of mitochondrial DNA in atherosclerotic lesions of human aortic intima. Biomolecules 2019;9:455.
40. Markin AM, Sobenin IA, Grechko AV, Zhang D, Orekhov AN. Cellular mechanisms of human atherogenesis: focus on chronification of inflammation and mitochondrial mutations. Front Pharmacol 2020;11:642.
41. Bezsonov EE, Sobenin IA, Orekhov AN. Immunopathology of atherosclerosis and related diseases: focus on molecular biology. Int J Mol Sci 2021;22:4080.
42. Mushenkova NV, Bezsonov EE, Orekhova VA, Popkova TV, Starodubova AV, Orekhov AN. Recognition of oxidized lipids by macrophages and its role in atherosclerosis development. Biomedicines 2021;9:915.
43. Mezentsev A, Bezsonov E, Kashirskikh D, Baig MS, Eid AH, Orekhov A. Proatherogenic sialidases and desialylated lipoproteins: 35 years of research and current state from bench to bedside. Biomedicines 2021;9:600.
44. Sobenin IA, Salonen JT, Zhelankin AV, et al. Low density lipoprotein-containing circulating immune complexes: role in atherosclerosis and diagnostic value. Biomed Res Int 2014;2014:205697.
45. Myasoedova VA, Kirichenko TV, Melnichenko AA, et al. Anti-atherosclerotic effects of a phytoestrogen-rich herbal preparation in postmenopausal women. Int J Mol Sci 2016;17:1318.
46. Malekmohammad K, Bezsonov EE, Rafieian-Kopaei M. Role of lipid accumulation and inflammation in atherosclerosis: focus on molecular and cellular mechanisms. Front Cardiovasc Med 2021;8:707529.
47. Puteri MU, Azmi NU, Kato M, Saputri FC. PCSK9 promotes cardiovascular diseases: recent evidence about its association with platelet activation-induced myocardial infarction. Life (Basel) 2022;12:190.
48. Ding Z, Liu S, Wang X, et al. Cross-talk between pcsk9 and damaged mtDNA in vascular smooth muscle cells: role in apoptosis. Antioxid Redox Signal 2016;25:997-1008.
49. Mahboobnia K, Pirro M, Marini E, et al. PCSK9 and cancer: rethinking the link. Biomed Pharmacother 2021;140:111758.
50. Mitrofanov KY, Zhelankin AV, Shiganova GM, et al. Analysis of mitochondrial DNA heteroplasmic mutations A1555G, C3256T, T3336C, С5178А, G12315A, G13513A, G14459A, G14846А and G15059A in CHD patients with the history of myocardial infarction. Exp Mol Pathol 2016;100:87-91.
51. Hefti E, Blanco JG. Mitochondrial DNA heteroplasmy in cardiac tissue from individuals with and without coronary artery disease. Mitochondrial DNA A DNA Mapp Seq Anal 2018;29:587-93.
52. Elsharawy MA, Alkhadra AH, Ibrahim MF, et al. Impact of atherosclerosis risk factors on the clinical presentation of arterial occlusive disease in Arabic patients. Int J Angiol 2008;17:203-6.
53. Vilela EM, Fontes-Carvalho R. Inflammation and ischemic heart disease: The next therapeutic target? Rev Port Cardiol (Engl Ed) 2021;40:785-96.
54. Linton MF, Yancey PG, Davies SS, et al. The role of lipids and lipoproteins in atherosclerosis. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000.
55. Liu YX, Yuan PZ, Wu JH, Hu B. Lipid accumulation and novel insight into vascular smooth muscle cells in atherosclerosis. J Mol Med (Berl) 2021;99:1511-26.
56. Hilgendorf I, Swirski FK, Robbins CS. Monocyte fate in atherosclerosis. Arterioscler Thromb Vasc Biol 2015;35:272-9.
58. Gupta RM, Lee-Kim VS, Libby P. The march of monocytes in atherosclerosis: one cell at a time. Circ Res 2020;126:1324-6.
59. Chistiakov DA, Orekhov AN, Sobenin IA, Bobryshev YV. Plasmacytoid dendritic cells: development, functions, and role in atherosclerotic inflammation. Front Physiol 2014;5:279.
60. Chistiakov DA, Sobenin IA, Orekhov AN, Bobryshev YV. Myeloid dendritic cells: Development, functions, and role in atherosclerotic inflammation. Immunobiology 2015;220:833-44.
61. Chistiakov DA, Sobenin IA, Orekhov AN. Strategies to deliver microRNAs as potential therapeutics in the treatment of cardiovascular pathology. Drug Deliv 2012;19:392-405.
62. Soldatov VO, Malorodova TN, Balamutova TI, Ksenofontov AO, Dovgan AP, Urozhevskaya ZS. Endothelial dysfunction: comparative evaluation of ultrasound dopplerography, laser dopplerflowmetry and direct monitoring of arterial pressure for conducting pharmacological tests in rats. Research Results in Pharmacology 2018;4:73-80.
63. Scioli MG, Storti G, D'Amico F, et al. Oxidative stress and new pathogenetic mechanisms in endothelial dysfunction: potential diagnostic biomarkers and therapeutic targets. J Clin Med 2020;9:1995.
64. Rodger CE, McWilliams TG, Ganley IG. Mammalian mitophagy - from in vitro molecules to in vivo models. FEBS J 2018;285:1185-202.
65. Iorio R, Celenza G, Petricca S. Mitophagy: molecular mechanisms, new concepts on parkin activation and the emerging role of AMPK/ULK1 axis. Cells 2021;11:30.
66. Vincow ES, Merrihew G, Thomas RE, et al. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci USA 2013;110:6400-5.
67. Kondapalli C, Kazlauskaite A, Zhang N, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2012;2:120080.
68. Lee L, Seager R, Nakamura Y, Wilkinson KA, Henley JM. Parkin-mediated ubiquitination contributes to the constitutive turnover of mitochondrial fission factor (Mff). PLoS One 2019;14:e0213116.
69. Koyano F, Yamano K, Kosako H, Tanaka K, Matsuda N. Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL. J Biol Chem 2019;294:10300-14.
70. Vives-Bauza C, de Vries RL, Tocilescu M, Przedborski S. PINK1/Parkin direct mitochondria to autophagy. Autophagy 2010;6:315-6.
71. Lazarou M. Keeping the immune system in check: a role for mitophagy. Immunol Cell Biol 2015;93:3-10.
72. Stockum S, Marchesan E, Ziviani E. Mitochondrial quality control beyond PINK1/Parkin. Oncotarget 2018;9:12550-1.
73. Fu M, St-Pierre P, Shankar J, Wang PT, Joshi B, Nabi IR. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell 2013;24:1153-62.
74. Ikeda F. Mitophagy is induced by short ubiquitin chains on mitochondria. J Cell Biol 2020;219:e202008031.
75. Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol 2018;20:233-42.
76. Montava-Garriga L, Ganley IG. Outstanding questions in mitophagy: what we do and do not know. J Mol Biol 2020;432:206-30.
77. Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 2013;15:1197-205.
78. Chu CT, Bayır H, Kagan VE. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: implications for Parkinson disease. Autophagy 2014;10:376-8.
79. Pizzuto M, Pelegrin P. Cardiolipin in immune signaling and cell death. Trends Cell Biol 2020;30:892-903.
80. Singh F, Prescott AR, Rosewell P, Ball G, Reith AD, Ganley IG. Pharmacological rescue of impaired mitophagy in Parkinson’s disease-related LRRK2 G2019S knock-in mice. Elife 2021;10:e67604.
81. McWilliams TG, Prescott AR, Montava-Garriga L, et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab 2018;27:439-449.e5.
82. Cummins N, Götz J. Shedding light on mitophagy in neurons: what is the evidence for PINK1/Parkin mitophagy in vivo? Cell Mol Life Sci 2018;75:1151-62.
83. Lee JJ, Sanchez-Martinez A, Martinez Zarate A, et al. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol 2018;217:1613-22.
84. Matsuda S, Nakanishi A, Minami A, Wada Y, Kitagishi Y. Functions and characteristics of PINK1 and Parkin in cancer. Front Biosci (Landmark Ed) 2015;20:491-501.
85. Quinn PMJ, Moreira PI, Ambrósio AF, Alves CH. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol Commun 2020;8:189.
86. Ge P, Dawson VL, Dawson TM. PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson’s disease. Mol Neurodegener 2020;15:20.
87. Li J, Xu X, Huang H, et al. Pink1 promotes cell proliferation and affects glycolysis in breast cancer. Exp Biol Med (Maywood) 2022;247:985-95.
88. McWilliams TG, Prescott AR, Allen GF, et al. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 2016;214:333-45.
89. Whitworth AJ, Pallanck LJ. PINK1/Parkin mitophagy and neurodegeneration-what do we really know in vivo? Curr Opin Genet Dev 2017;44:47-53.
90. Sekine S, Youle RJ. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol 2018;16:2.
91. Lin XH, Qiu BQ, Ma M, et al. Suppressing DRP1-mediated mitochondrial fission and mitophagy increases mitochondrial apoptosis of hepatocellular carcinoma cells in the setting of hypoxia. Oncogenesis 2020;9:67.
92. Schweers RL, Zhang J, Randall MS, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A 2007;104:19500-5.
93. Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008;454:232-5.
94. Al Rawi S, Louvet-Vallée S, Djeddi A, et al. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011;334:1144-7.
95. Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 2011;334:1141-4.
96. Rojansky R, Cha MY, Chan DC. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife 2016;5:e17896.
97. Gottlieb RA, Stotland A. MitoTimer: a novel protein for monitoring mitochondrial turnover in the heart. J Mol Med (Berl) 2015;93:271-8.
98. Esteban-Martínez L, Sierra-Filardi E, Boya P. Mitophagy, metabolism, and cell fate. Mol Cell Oncol 2017;4:e1353854.
99. Suárez-Rivero JM, Pastor-Maldonado CJ, Povea-Cabello S, et al. From mitochondria to atherosclerosis: the inflammation path. Biomedicines 2021;9:258.
100. Summerhill VI, Grechko AV, Yet SF, Sobenin IA, Orekhov AN. The atherogenic role of circulating modified lipids in atherosclerosis. Int J Mol Sci 2019;20:3561.
101. Georgakopoulos ND, Wells G, Campanella M. The pharmacological regulation of cellular mitophagy. Nat Chem Biol 2017;13:136-46.
102. Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012;8:1462-76.
103. Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010;8:e1000298.
104. Bestman JE, Stackley KD, Rahn JJ, Williamson TJ, Chan SS. The cellular and molecular progression of mitochondrial dysfunction induced by 2,4-dinitrophenol in developing zebrafish embryos. Differentiation 2015;89:51-69.
105. Geisler JG, Marosi K, Halpern J, Mattson MP. DNP, mitochondrial uncoupling, and neuroprotection: a little dab’ll do ya. Alzheimers Dement 2017;13:582-91.
106. Tai Y, Li L, Peng X, et al. Mitochondrial uncoupler BAM15 inhibits artery constriction and potently activates AMPK in vascular smooth muscle cells. Acta Pharm Sin B 2018;8:909-18.
107. Dang CP, Issara-Amphorn J, Charoensappakit A, et al. BAM15, a mitochondrial uncoupling agent, attenuates inflammation in the lps injection mouse model: an adjunctive anti-inflammation on macrophages and hepatocytes. J Innate Immun 2021;13:359-75.
108. Felber SM, Brand MD. Valinomycin can depolarize mitochondria in intact lymphocytes without increasing plasma membrane potassium fluxes. FEBS Letters 1982;150:122-4.
109. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2013;20:31-42.
110. Rakovic A, Ziegler J, Mårtensson CU, et al. PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ 2019;26:1428-41.
111. Kim KY, Yu SN, Lee SY, et al. Salinomycin-induced apoptosis of human prostate cancer cells due to accumulated reactive oxygen species and mitochondrial membrane depolarization. Biochem Biophys Res Commun 2011;413:80-6.
112. Managò A, Leanza L, Carraretto L, et al. Early effects of the antineoplastic agent salinomycin on mitochondrial function. Cell Death Dis 2015;6:e1930.
113. Thomas RE, Andrews LA, Burman JL, Lin WY, Pallanck LJ. PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genet 2014;10:e1004279.
114. Shi G, Lee JR, Grimes DA, et al. Functional alteration of PARL contributes to mitochondrial dysregulation in Parkinson’s disease. Hum Mol Genet 2011;20:1966-74.
115. Meissner C, Lorenz H, Hehn B, Lemberg MK. Intramembrane protease PARL defines a negative regulator of PINK1- and PARK2/Parkin-dependent mitophagy. Autophagy 2015;11:1484-98.
116. Shi G, McQuibban GA. The mitochondrial rhomboid protease PARL is regulated by PDK2 to integrate mitochondrial quality control and metabolism. Cell Rep 2017;18:1458-72.
117. Lysyk L, Brassard R, Arutyunova E, et al. Insights into the catalytic properties of the mitochondrial rhomboid protease PARL. J Biol Chem 2021;296:100383.
118. Urban S, Dickey SW. The rhomboid protease family: a decade of progress on function and mechanism. Genome Biol 2011;12:231.
119. Parsons WH, Rutland NT, Crainic JA, et al. Development of succinimide-based inhibitors for the mitochondrial rhomboid protease PARL. Bioorg Med Chem Lett 2021;49:128290.
120. Hertz NT, Berthet A, Sos ML, et al. A neo-substrate that amplifies catalytic activity of parkinson's-disease-related kinase PINK1. Cell 2013;154:737-47.
121. Clark EH, Vázquez de la Torre A, Hoshikawa T, Briston T. Targeting mitophagy in Parkinson’s disease. J Biol Chem 2021;296:100209.
122. Nagaria O, Singh S, Kabir R, Kobayashi S, Kobayashi T, Liang Q. Kinetin is sufficient to accelerate mitophagy flux in H9c2 cardiac myoblast cells. FASEB j 2019:33.
123. Allen GF, Toth R, James J, Ganley IG. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep 2013;14:1127-35.
124. Lee JW, Ko J, Ju C, Eltzschig HK. Hypoxia signaling in human diseases and therapeutic targets. Exp Mol Med 2019;51:1-13.
125. Zhao JF, Rodger CE, Allen GFG, Weidlich S, Ganley IG. HIF1α-dependent mitophagy facilitates cardiomyoblast differentiation. Cell Stress 2020;4:99-113.
126. Park SJ, Shin JH, Kim ES, et al. Mitochondrial fragmentation caused by phenanthroline promotes mitophagy. FEBS Lett 2012;586:4303-10.
127. Hara Y, Yanatori I, Tanaka A, et al. Iron loss triggers mitophagy through induction of mitochondrial ferritin. EMBO Rep 2020;21:e50202.
128. Xu LJ, Jin L, Pan H, et al. Deferiprone protects the isolated atria from cardiotoxicity induced by doxorubicin. Acta Pharmacol Sin 2006;27:1333-9.
129. Denison SR, Wang F, Becker NA, et al. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene 2003;22:8370-8.
130. Long M, Sanchez-Martinez A, Longo M, et al. DGAT1 activity synchronises with mitophagy to protect cells from metabolic rewiring by iron depletion. EMBO J 2022;41:e109390.
131. Read AD, Bentley RE, Archer SL, Dunham-Snary KJ. Mitochondrial iron-sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox Biol 2021;47:102164.
132. Paul BT, Manz DH, Torti FM, Torti SV. Mitochondria and Iron: current questions. Expert Rev Hematol 2017;10:65-79.
133. Ward DM, Cloonan SM. Mitochondrial iron in human health and disease. Annu Rev Physiol 2019;81:453-82.
136. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia 2017;60:1577-85.
137. Bhansali S, Bhansali A, Dhawan V. Metformin promotes mitophagy in mononuclear cells: a potential in vitro model for unraveling metformin’s mechanism of action. Ann N Y Acad Sci 2020;1463:23-36.
138. Pan H, Finkel T. Key proteins and pathways that regulate lifespan. J Biol Chem 2017;292:6452-60.
139. Jang SY, Kang HT, Hwang ES. Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J Biol Chem 2012;287:19304-14.
140. Sebori R, Kuno A, Hosoda R, Hayashi T, Horio Y. Resveratrol decreases oxidative stress by restoring mitophagy and improves the pathophysiology of dystrophin-deficient mdx mice. Oxid Med Cell Longev 2018;2018:9179270.
141. Shaito A, Posadino AM, Younes N, et al. Potential adverse effects of resveratrol: a literature review. Int J Mol Sci 2020;21:2084.
143. Jia S, Xu X, Zhou S, Chen Y, Ding G, Cao L. Fisetin induces autophagy in pancreatic cancer cells via endoplasmic reticulum stress- and mitochondrial stress-dependent pathways. Cell Death Dis 2019;10:142.
144. Hung CH, Chan SH, Chu PM, Tsai KL. Quercetin is a potent anti-atherosclerotic compound by activation of SIRT1 signaling under oxLDL stimulation. Mol Nutr Food Res 2015;59:1905-17.
145. Li X, Wang H, Gao Y, et al. Protective effects of quercetin on mitochondrial biogenesis in experimental traumatic brain injury via the Nrf2 signaling pathway. PLoS One 2016;11:e0164237.
146. Liu T, Yang Q, Zhang X, et al. Quercetin alleviates kidney fibrosis by reducing renal tubular epithelial cell senescence through the SIRT1/PINK1/mitophagy axis. Life Sci 2020;257:118116.
147. Pietrocola F, Castoldi F, Markaki M, et al. Aspirin recapitulates features of caloric restriction. Cell Rep 2018;22:2395-407.
148. Eisenberg T, Knauer H, Schauer A, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 2009;11:1305-14.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Borisov E, Bezsonov E, Lyukmanov D, Poggio P, Moschetta D, Valerio V. Pharmacological agents affecting mitophagy and inflammation. Vessel Plus 2022;6:63. http://dx.doi.org/10.20517/2574-1209.2022.20
AMA Style
Borisov E, Bezsonov E, Lyukmanov D, Poggio P, Moschetta D, Valerio V. Pharmacological agents affecting mitophagy and inflammation. Vessel Plus. 2022; 6: 63. http://dx.doi.org/10.20517/2574-1209.2022.20
Chicago/Turabian Style
Borisov, Evgeny, Evgeny Bezsonov, Damir Lyukmanov, Paolo Poggio, Donato Moschetta, Vincenza Valerio. 2022. "Pharmacological agents affecting mitophagy and inflammation" Vessel Plus. 6: 63. http://dx.doi.org/10.20517/2574-1209.2022.20
ACS Style
Borisov, E.; Bezsonov E.; Lyukmanov D.; Poggio P.; Moschetta D.; Valerio V. Pharmacological agents affecting mitophagy and inflammation. Vessel Plus. 2022, 6, 63. http://dx.doi.org/10.20517/2574-1209.2022.20
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 21 clicks
Cite This Article 21 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.