
A novel insight into the nature of modified low-density lipoproteins and their role in atherosclerosis

 , ...
, ... Abstract
Atherosclerosis plays a significant role in the development of cardiovascular diseases, the leading cause of death worldwide. Modification of low-density lipoproteins (LDLs) is a critical event in atherogenesis. Native LDL undergoes several modifications that can lead to the formation of atherogenic modified LDLs. LDL modifications change their physicochemical and biological properties. Possible modifications include changes in the lipoprotein particle’s structure, size, charge, and composition. Uptake and utilization of modified LDLs are impaired in cells. Macrophages take up modified LDLs that promote forming of foam cells, one of the critical cellular components of atherosclerotic lesions. Nevertheless, the direct role of each atherogenic LDL modification in atherogenesis remains uncertain. This review highlights LDL's most critical atherogenic modifications, including oxidized, enzyme-modified, non-oxidative, desialylated, glycated and carbamylated LDLs. Studying the role of each type of LDL modification will clarify the unknown elements of atherosclerosis progression and facilitate the development of effective methods for its diagnosis, treatment, and prevention.
Keywords
INTRODUCTION
According to the World Health Organization, cardiovascular disease (CVD) is the leading cause of morbidity and mortality worldwide, representing 32% of all deaths. Atherosclerosis is associated with the occurrence and development of CVDs, including coronary heart disease (CHD), myocardial infarction, stroke, and peripheral arterial disease. The study of atherosclerosis initiation and early development would make it possible to develop practical diagnostic tools for the asymptomatic stages and promising therapeutic approaches[1].
Atherosclerosis is a chronic progressive disease of the elastic arteries characterized by the accumulation and retention of cholesterol ApoB-containing lipoproteins, primarily low-density lipoproteins (LDLs), in the arterial wall[2]. The pathogenic mechanism of atherosclerosis is a cascade of events leading to the formation of atherosclerotic lesions. Currently, there are disputes about the processes that initiate atherosclerosis. The critical events in the formation of atherosclerotic lesions are changes in endothelial permeability, migration of smooth muscle cells, synthesis of extracellular matrix components, retention of lipoproteins in the vessel wall, modification of lipoproteins, turbulent blood flow, and inflammation[3-6]. This review is focused on the modification of LDL. One of the critical events of atherogenesis is LDL modification[4,7,8]. It has been repeatedly demonstrated that modified LDL causes excessive intracellular accumulation of lipids in cell cultures, which determines lipoprotein atherogenicity[9-12]. It has been shown that incubating culture of human aortic intima cells with native LDL does not lead to intracellular lipid accumulation[12,13]. Modified LDLs can be found in atherosclerotic lesions and in the blood of patients with atherosclerosis[14-17]. Circulating LDLs in patients with atherosclerosis CHD are modified several times, and modified LDLs may differ in terms of the modification from one patient to another[18-20]. Such features of LDL are likely to be associated with various combinations of modifications[13,20]. Modified LDLs can be classified as oxidized LDL (OxLDL), enzyme-modified non-oxidized LDL (eLDL), desialylated LDL (desLDL), glycated LDL (gLDL), and carbamylated LDL (cLDL) [Figure 1].
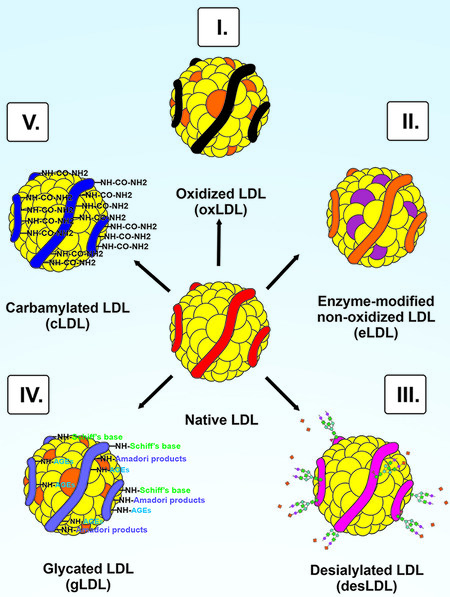
Figure 1. The scheme of LDL modifications. (I) Oxidation of native LDL with reactive oxygen species, reactive nitrogen species, cyclooxygenase 2, lipoxygenases, cytochrome P450 monooxygenases, myeloperoxidase, and lipoxygenase 12/15. (II) Enzymatic non-oxidative modification of native LDL by matrix metalloproteases 2 and 9, plasmin, trypsin, cholesterol esterase, chymases, sphingomyelinase, phospholipase A2, and cathepsins (cathepsin H, G, and F). (III) Desialylation of native LDL by endogenous sialidases, trans-sialidase, viral neuraminidases, and reactive oxygen species. (IV) Glycation of native LDL by excess glucose and nornicotine. (V) Carbamylation of native LDL with urea, cyanate, and thiocyanate with myeloperoxidase and H2O2.
There is continuing interest in studying LDL modifications and their role in atherogenesis. Over the past five years, 13,758 full-text articles with the keywords “modified LDL and atherosclerosis” have been registered in the PubMed Central system. These articles account for about half of all publications that mention a combination of these terms from 1979 to 2022. However, several LDL modifications in atherogenesis remain incompletely elucidated. Therefore, this review highlights the role of various mechanisms of LDL modifications in the early stages of atherosclerosis development.
OXIDIZED LDL
The oxidative modification hypothesis
The oxidative modification hypothesis arose from the study of atherosclerosis in patients with homozygous hypercholesterolemia. Although this disease is characterized by the absence of expression of the LDL receptor (LDLR), foam cell formation is observed in patients with homozygous hypercholesterolemia[21]. It is essential to note that LDLR has usually involved in LDL receptor-mediated endocytosis[22]. In 1979, Goldstein and Brown suggested that LDL modification occurs before the LDL uptake by macrophages with subsequent transformation into foam cells[21]. In addition, it was assumed that an alternative receptor is required to uptake modified LDLs. Goldstein and Brown showed that adding chemically acetylated LDL (acLDL) to cell culture led to their uptake and more significant intracellular cholesterol levels than native LDL[23]. The acLDL receptor was characterized and named scavenger receptor A (SR-A)[24]. Goldstein and Brown demonstrated the participation of modified LDL in atherogenesis; however, acLDL has never been found in vivo; therefore, a search is required to establish new modified LDLs involved in atherogenesis[21].
Developing the idea of the participation of modified LDL in atherogenesis, Steinbrecher and Hessler suggested that free radical lipids are involved in LDL modification[25,26]. Incubation of native LDL with endothelial cells and human dermal fibroblasts results in LDL oxidation by lipid peroxidation (LPO)[25,26]. The evidence supports the oxidative modification hypothesis of LDL: (1) there is OxLDL in vivo; (2) OxLDL leads to the formation of foam cells compared to native LDL; and (3) high levels of ROS and their products present in atherosclerotic lesions[18,27-29]. In addition, the predictive value of serum OxLDL levels in determining the progression of subclinical atherosclerosis has recently been demonstrated[30].
There are several current assumptions regarding the moment when LDL oxidation occurs: before or after macrophage uptake. According to the response-to-retention hypothesis, native LDL accumulates in the vessel wall[3]. One explanation for the excess accumulation of lipids is a change in the length of chondroitin sulfate chains in the tunica intima[31,32]. In addition, changes in the length of macrophage chondroitin sulfate proteoglycans have been shown to affect OxLDL binding on the surface of peritoneal macrophages[33,34]. According to the oxidative modification hypothesis, atherosclerosis is initiated by LDL oxidation in the vascular wall[35]. Moreover, it is essential to note that OxLDL in the blood was excluded due to several facts: (1) LDL contains an antioxidant component (coenzyme-Q10, α-tocopherol, and carotenoids); and (2) high activity of the antioxidant system in the blood[4,36]. LDL oxidation can be divided into two successive stages[4]. In the first stage, the lipid part of LDL is oxidized, while the protein part is almost not modified. In the second stage, the formed mildly oxidized LDL undergoes strong oxidation of the LDL protein part, leading to the formation of highly oxidized LDL[37]. Modifying ApoB-100 promotes increased uptake of OxLDL by macrophages and subsequent formation of foam cells[23,38]. Thus, forming foam cells is a pathological characteristic of atherosclerosis in the early stages[39].
The lysosomal pathway is considered the second possible pathway of LDL oxidation[40]. Aggregated LDL can be oxidized in macrophage lysosomes in the presence of iron[41]. Acidic pH is required for cholesterol esterase (CEase) activity and proteases for LDL degradation in normal conditions. Incubation of THP-1 macrophages with LDL aggregated by sphingomyelinase (SMase-LDL) leads to an increase in pH and a decrease in the degradation of endocytosed LDL[42]. The lysosome-targeted antioxidant cysteamine can prevent intracellular LDL accumulation and oxidation. Cysteamine prevents an increase in pH in lysosomes, maintaining the proteolytic activity of lysosomes and reducing LDL oxidation[42]. The mechanism of lysosomal LDL oxidation likely explains the failure of common antioxidants in clinical trials to reduce free radical processes[42,43].
It remains unclear whether oxidative stress is a cause or a consequence of atherosclerosis. However, lipoprotein oxidation is thought to be one of the processes present in the early stages of atherosclerosis[4,7]. In addition, according to recent data, OxLDL is a risk factor for CVD and dyslipidemia[44].
Mechanism of LDL oxidation
The oxidation process involves transferring electrons from an electron donor to an electron acceptor. LDL oxidation can be carried out both with the participation of specialized enzymes (cyclooxygenases, lipoxygenases, cytochrome P450 monooxygenases, and myeloperoxidase) and without them (metals with variable valences)[45-48]. Moreover, LDL’s protein and lipid parts are prone to oxidation[26] [Figure 2]. LPO products can form Schiff bases, and Michael adducts with ApoB-100 residues. Oxidation of the LDL protein part leads to impaired recognition of OxLDL by LDLR, increasing the circulating lifetime of LDL. Moreover, OxLDL undergoes subsequent alternative binding to scavenger receptors (SR)[15]. The primary components of the LDL lipid part, such as cholesterol esters (CE), phospholipids (PL), and triglycerides, undergo LPO, followed by the formation of hydroperoxides and aldehyde derivatives (malonic dialdehyde and 4-hydroxynonenal[10,49,50]. Furthermore, there is a clear relationship between the degree of LPO and polyunsaturated fatty acids of LDL[51]. Well-known polyunsaturated fatty acids such as arachidonic acid (AA), eicosapentaenoic acid, and linoleic acid also undergo oxidative modification. The lipoxygenase 12/15 (12/15-LOX) carries out oxidative modification of LDL direct oxygenation and LRP-mediated 12/15-LOX membrane translocation. In the former case, 12/15-LOX oxygenates esterified fatty acids in lipoproteins and phospholipids with the formation of biologically active compounds[52,53]; as a result, 12-hydroperoxyeicosatetraenoic acid (12-HPETE) and 15-HPETE from AA and 13-hydroperoxyoctadecadienoic acid (13-HPETE) from linoleic acid are formed[54,55]. In the latter case, it was shown that 12/15-LOX moves from the cytoplasm to the plasma membrane and oxidizes LDL during pre-incubation of macrophages with LDL[56]. This process is closely related to the receptor-related protein (LRP)[57]. However, the exact mechanism of LRP-mediated 12/15-LOX membrane translocation remains unclear[58]. 12/15-LOX has been reported to be directly involved in OxLDL formation[59,60]. 12/15-LOX has been shown to have a pro-atherogenic role by affecting the area of atherosclerotic lesions and the sensitivity of LDL to oxidation in transgenic mice[46,61]. Importantly, lipoxygenase-15 has been found in atherosclerotic lesions where it can colocalize with OxLDL[62,63].
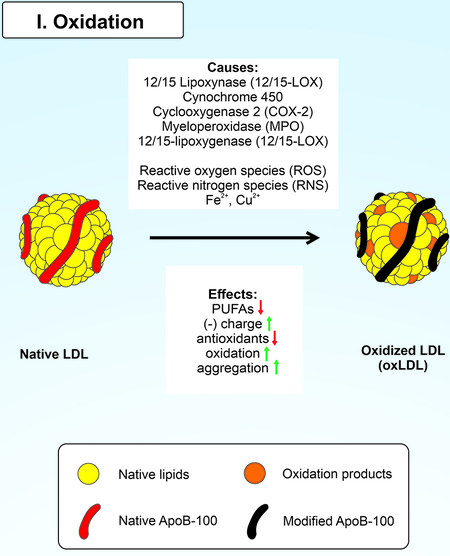
Figure 2. Oxidation of native LDL.
Cyclooxygenases catalyze the oxygenation of AA to prostaglandins (PGs)[64]. PGs, as a consequence, are rapidly converted to PGE2, PGD2, PGF2, and PGJ2, which initiate many of the responses associated with inflammation and vascular reactivity[65]. In addition, there is evidence of pro-atherogenic and anti-atherogenic effects of cyclooxygenase 2 (COX-2)[45,66]. The various biological effects of COX-2 products are determined by the type of tissue in which they are produced[66]. Cytochrome P450 monooxygenases catalyze the conversion of AAs to epoxyeicosatrienoic acids[64,67], which regulate vascular tone and are associated with anti-inflammatory effects[68-70].
Another enzyme responsible for LDL oxidation is MPO[71]. It has been reported that there is an association between serum MPO levels and acute coronary syndrome[72,73]. MPO forms a range of reactive compounds such as hypochlorous acid (HOCl), chloramines, tyrosyl radicals, and nitrogen dioxide (NO2) that oxidize the protein, lipid, and antioxidant components of LDL[47]. In addition, myeloperoxidase-oxidized LDL (Mox-LDL) is found in atherosclerotic lesions and in the blood of patients with atherosclerosis[14,15].
Metals with variable valences, such as iron and copper ions, are involved in LDL oxidation by interacting with lipid hydroperoxides[48]. Moreover, there is evidence of the participation of ROS in LDL oxidation by forming hydroxyl radicals from hydrogen peroxide and interacting with superoxide radicals[74,75]. It is essential to note that the primary enzymatic sources of ROS are NADPH oxidase, endothelial nitric oxide synthase (eNOS), and xanthine oxidase (XO)[76]. Likely, the generation of reactive nitrogen species such as peroxynitrite may contribute to LDL oxidative modification[47].
Effects of oxidized LDL
The oxidation process of native LDL leads to the appearance of pro-atherogenic properties in modified LDLs. Oxidative modification of the protein part impairs LDL recognition by LDLR. Macrophages actively take up OxLDL by scavenger receptors (CD36, SR-A, and SR-B1) and lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) to remove excess OxLDL from the arterial wall[77-79]. OxLDL may be involved in ROS production, leading to endothelial cell damage[80]. OxLDL stimulates monocyte adhesion and triggers the differentiation of intimal monocytes into resident macrophages[81]. Furthermore, OxLDL induces the production of pro-inflammatory cytokines and chemokines by monocytes, endothelial cells, and smooth muscle cells (SMCs)[82]. In addition, OxLDL inhibits autophagy, a cellular pathway for the degradation of organelles and macromolecules. Autophagy leads to excessive accumulation of intracellular lipids and endothelial cell apoptosis[11]. Thus, excessive uptake of OxLDL by macrophages leads to lipid accumulation followed by foam cell formation, which is the hallmark of the early stages of atherosclerosis[83]. Mox-LDL causes endothelial dysfunction through the uptake by LOX-1, contributing to the initiation or development of atherosclerosis[14]. The use of substances lowering the uptake of OxLDL, such as dioscin and nifedipine, can reduce the development of atherosclerosis[84,85].
As mentioned earlier, these lines of evidence support the involvement of OxLDL in atherosclerosis in the early stages. However, most of the experiments were carried out using cell cultures, so further studies in animal models are required.
ENZYME-MODIFIED NON-OXIDIZED LDL
Mechanism of enzymatic non-oxidative modification of native LDL
According to the hypothesis of the enzymatic non-oxidative LDL modification, native LDLs infiltrate the intima and are enzymatically modified[86]. Some enzymes can modify LDL in vitro without oxidation processes. Such modified LDL particles are called enzyme-modified non-oxidized LDL (eLDL) [Figure 3]. Enzymatic non-oxidative modification of LDL occurs due to the activity of ubiquitous hydrolytic enzymes such as matrix metalloproteases 2 and 9, plasmin, trypsin, CEase, chymases, sphingomyelinase (SMase), phospholipase A2 (PLA2) and cathepsins H, G and F[16,42,87-92]. eLDL is prone to aggregation and fusion, increasing lipoprotein retention by human aortic proteoglycans due to active ApoB-100 lysine residues[92]; eLDL phagocytosed by macrophages is transported to lysosomes, where it can be oxidized[42].
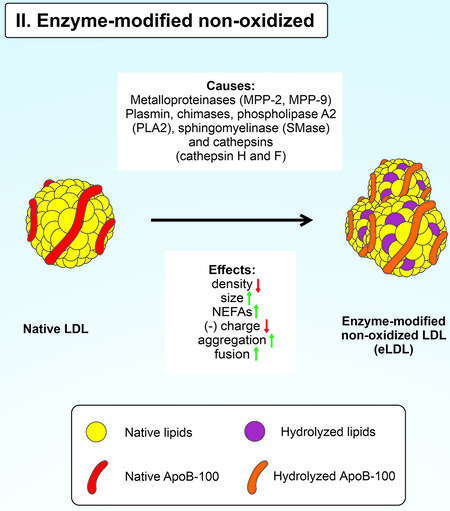
Figure 3. Enzymatic non-oxidative modification of native LDL.
Hydrolytic enzymes affect the protein and lipid parts of LDL. Plasmin can induce fragmentation of LDL ApoB-100, while trypsin and cathepsins proteolyze the protein part of LDL, followed by the formation of aggregates from its fragments[90,93]. Lipolytic modifications of LDL are carried out by lipases such as SMase and PLA2. SMase catalyzes the hydrolysis of sphingomyelins by forming ceramides and phosphorylcholines[91]. Moreover, eLDL has a lower density and exhibits less electrophoretic mobility than native LDL, indicating its lower negative charge[93,94]. PLA2 degrades LDL phospholipids to form non-esterified fatty acids and lysophospholipids[95]. Treatment of native LDL with SMase leads to aggregation and fusion of particles, while incubation of LDL with PLA2 leads only to aggregation[92]. Thus, this event leads to their increased uptake by macrophages[96]. There is also a lipoprotein-associated PLA2 that preferentially hydrolyzes oxidized phospholipids of OxLDL [97]. Human aortic proteoglycans actively retain such an eLDL[86,92].
The LDL core lipids, such as cholesteryl esters and triglycerides, may undergo hydrolysis. CEase catalyzes the conversion of cholesterol ester to non-esterified cholesterol and fatty acid. CE hydrolysis leads to the formation of cholesterol crystals found in early and advanced atherosclerotic lesions[98-100]. Furthermore, eLDL induces intracellular lipid accumulation by cells and promotes the formation of lipid droplets[10].
It is most likely that native LDL undergoes lipolytic and proteolytic modifications. It has been shown that the combined treatment of native LDL with trypsin, Cease, and neuraminidase leads to the formation of eLDL with characteristics similar to the eLDL from atherosclerotic lesions[86,101].
Effects of enzyme-modified non-oxidative LDL
It is essential to note that eLDL has pro-atherogenic properties. It is actively taken up by SMCs of coronary arteries and macrophages derived from monocytes[101]. Uptake of eLDL has been shown to occur via micropinocytosis and is independent of scavenger receptors[94]. Nevertheless, eLDL activates the major OxLDL scavenger receptor LOX-1 in SMCs[94]. Thus, eLDL aggregation and fusion have been shown to promote eLDL uptake by macrophages and foam cell formation[42]. There is evidence of an association between the aggregation-susceptibility of LDL particles and future cardiovascular events in patients with atherosclerosis[102,103].
There is evidence of the association of eLDL with inflammatory responses in atherosclerosis. Thus, eLDL activates the complement system in a C-reactive protein-dependent fashion[104,105]. Other potential triggers for the complement system activation in the arterial wall include modified lipoproteins and cholesterol crystals[106,107]. Modified and aggregated eLDL activate macrophages and induce the secretion of pro-inflammatory cytokines and chemokines. Incubation of THP-1-derived macrophages with SMase-LDL induces secretion of TNF, IL-1β, IL-6, and MCP-1[42,98]. Treatment of native LDL with PLA2 or SMase leads to the generation of pro-inflammatory lipid mediators from AA[108].
eLDL triggers migration and osteogenic differentiation of SMCs and OxLDL[16,109,110]. The development of the advanced atherosclerotic lesion is characterized by plaque calcification and SMC migration from the media to the intima of the arterial wall. SMCs play an essential role in forming the protective fibrous cap; therefore, their dysfunction usually leads to plaque rupture[111]. eLDL has been found in atherosclerotic lesions and aortic valve sclerosis[16,112,113].
In general, eLDL is involved in the pathological processes that characterize early and advanced atherosclerotic lesions. Pharmacological reduction of SMase and PLA2 activity would decrease eLDL formation[114,115]. Moreover, using substances inhibiting macropinocytosis and eLDL aggregation is a promising approach for atherosclerosis treatment[16,116]. However, further research is needed on the many mechanisms of eLDL formation and their effects due to the ubiquitous presence of various hydrolytic enzymes in atherosclerotic lesions.
DESIALYLATED LDL
The desialylation modification hypothesis
Chazov et al. first reported serum atherogenicity in patients with CHD in 1986[117]. The addition of serum from patients with CHD to a primary culture of human aortic intima subendothelial cells increased intracellular lipid accumulation, in contrast to serum from healthy patients. The separation of lipoproteins into classes made it possible to identify LDL as an atherogenic serum component of CHD patients[118,119]. Further study of lipoproteins showed that atherogenic LDL is desialylated. This atherogenic LDL had decreased sialic acid level (by 2-5 times) compared to LDL isolated from non-atherogenic plasma. Furthermore, native LDL from healthy donors became atherogenic after incubation with neuraminidase from Clostridium perfringens[9]. Subsequently, a subfraction of desialylated LDL was isolated from the atherogenic serum of CHD patients using lectin blotting with Ricinus communis[120,121]. Moreover, multi-modified LDL is significantly different from native LDL; the densest subfractions of desialylated LDL caused intracellular lipid accumulation in cell culture[122]. Finally, the trans-sialidase, which transfers sialic acids from protein donors to other acceptors, has been isolated from human atherogenic serum[123]. The discovery of this enzyme and the results of subsequent studies led to conclusions about the possible effect of trans-sialidase on the initiation and development of atherosclerosis[124].
Despite these facts, there is a limited understanding of the nature of the desialylation process and its role in atherosclerosis. Further research on enzymatic and non-enzymatic desialylation mechanisms is required.
Sialylation
Sialylation is the glycosylation process of biological molecules such as glycolipids and glycoproteins. Sialylation plays a significant role in many biological processes, including embryonic development, reprogramming of somatic cells, immune responses, and oncogenesis[125-127]. It is essential to note that sialylation is carried out by sialyltransferase, which attaches activated cytidine-5'-monophosphate-sialic acid to the end of the O-glycan of serine or threonine or the terminal N-glycan of asparagine. Sialic acids are usually attached either to galactose or N-acetylgalactosamine residues of glycans at the α-2,3- or α-2,6-bond position or to other sialic acid moieties via α-2,8- or α-2,9 bonds.
Mechanism of LDL desialylation
Sialic acids can be cleaved from gangliosides by specific enzymes such as sialidases and without them. This process is called desialylation [Figure 4]. Desialylation and sialylation are essential parts of sialic acid metabolism[128]. However, desialylation is often associated with pathological processes such as sporadic prion disease, infectious diseases, and atherosclerosis[123,129,130]. Desialylation affects endothelial permeability in atherosclerosis. A recent study reported visualization of changes in the vascular endothelium after exposure to neuraminidase[131]. The enzyme cleaved the sialic acid residues, which reduced the depth of the endothelial glycocalyx and increased vascular permeability.
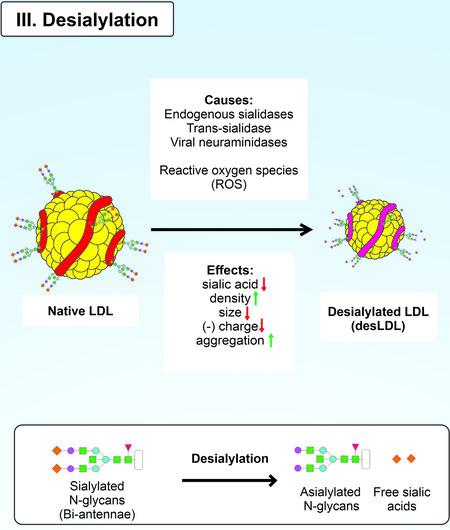
Figure 4. Desialylation of native LDL.
Desialylation may occur non-enzymatically. The formation of ROS can likely promote the cleavage of terminal sialic acids of glycans[132]. Previously, it was assumed that free radical processes are involved in LDL desialylation. Oxidation of LDL with copper ions led to a dose-dependent decrease in sialic acid content in vitro[133]. In another study, the sialic acid content on the cell surface decreased during the addition of hypoxanthine and xanthine oxidase (HX/XO) to HL60 cell culture[132]. This finding was explained by HX/XO causing the generation of a superoxide anion in the presence of metal ions of variable valence and hydrogen peroxide leading to the elimination of terminal sialic acid from glycosides. The cleavage of sialic acid from 4MU-Neu5Ac led to induced fluorescence. It is also essential to note that Neu5Ac exhibits antioxidant activity by neutralizing H2O2 with the formation of its oxidation product, 4-(acetylamino)-2,
Another example of the involvement of ROS in desialylation is a study on Long-Evans Cinnamon (LEC) rats, which are used to model hepatitis[135]. The inbred mutant strain serum samples were characterized by an increased level of hydrogen peroxide, copper, and LPO. In addition, serum glycolipids from LEC rats were desialylated compared to wild-type rats. During the exposure of healthy rats to copper ions and hydrogen peroxide, a decrease in trisialic acid chains and an increase in bi- and asialic acid chains of transferrin glycans were noted, indicating desialylation[135]. Another study using rabbits demonstrated increased levels of thiobarbituric acid reactive substances as atherosclerosis progressed, suggesting an increase in LPO[136]. Increased serum sialidase activity of serum and decreased sialic acid levels in the LDL fraction were also revealed in rabbits. Despite the evidence mentioned above about the mechanism of ROS-induced desialylation, further study is required.
Enzymatic desialylation is the most studied process of the cleavage of sialic acids from glycolipids and glycoproteins[137]. Sialidases (also known as neuraminidases) are a large group of enzymes. Hydrolytic sialidases and trans-sialidases belong to the same class of exo-alpha-sialidases (EC 3.2.1.18). Mammals have four neuraminidases: lysosomal (NEU1), cytosolic (NEU2), plasmatic (NEU3), and mitochondrial (NEU4)[138-141]. Hydrolytic sialidases can cleave α2-3-, α2-6-, and α2-8 terminal sialic acids from a wide range of biomolecules. Several studies demonstrated the role of endogenous neuraminidase in atherogenesis. Hypomorphic gene expression of Neu1 was shown to reduce inflammatory cell infiltration into vascular intima and decrease serum LDL cholesterol levels and atherosclerotic lesions size[142]. In another study, Neu1-deficient and Neu4-knockout Apoe-/-mice markedly slowed the development of atherosclerotic lesions compared to control Apoe-/- mice[143]. The use of endogenous sialidase inhibitors contributed to a decrease in the size of the atherosclerotic lesions in Apoe-/- mice[143]. These findings suggest that sialidase inhibition is a promising approach for atherosclerosis treatment[144].
The trans-sialidases may be responsible for LDL desialylation. Trans-sialidase, including protozoan trans-sialidase, is found in the blood of CHD patients, where it interacts with α2-3-terminal sialic acids, cleaving and transferring them to other glycolipids and glycoproteins[145,146]. As a sialic acid donor, this enzyme can use plasma lipoproteins, gangliosides, plasma protein glycoconjugates, and erythrocyte glycoconjugates. Erythrocyte glycoconjugates, plasma lipoproteins, and plasma proteins can be used as sialic acid acceptors. Sialic acid can be attached to terminal galactose, acetylgalactosamine, or other sialic acids at the α-2.3 or
The possible causes of atherogenic LDL modification could be viral and bacterial sialidases. A significant association has been found between influenza virus infection and an increased risk of acute myocardial infarction[147]. Furthermore, the influenza virus has been shown to aggravate OxLDL-induced endothelial cell apoptosis[148]. A seasonal pattern of the viral sialidase activity in the blood was also found, which might be an additional atherogenic factor[149]. Modeling of high sialidase activity in the blood by injection of Vibrio cholerae neuraminidase contributed to a reduced LDL sialic acid content in wild-type mice[8].
There are other enzymes with sialidase activity. The soluble form of the Klotho protein acts as a sialidase, removing terminal α-2,6-sialic acids from the N-glycans of the TRPV5 and ROMK1 ion channel protein chain[150,151]. This enzyme regulates cell surface glycoproteins’activity, affecting the balance of Ca2+ and K+ ions. It is essential to note that the Klotho protein has anti-atherogenic effects. This enzyme attenuates OxLDL-induced oxidative stress by activating the PI3K/Akt/eNOS pathway and downregulating LOX-1 expression[152]. There is also evidence of catalytic sialidase activity of abzymes. Abzymes desialylate molecules such as glycoproteins, gangliosides, and erythrocyte membranes, likely facilitating the clearance of apoptotic cells[153].
Effects of desialylated LDL
Desialylation is a critical atherogenic modification of LDL in the early stages of atherosclerosis[8]. An inverse correlation was found between LDL sialic acid content and lipoprotein atherogenicity[20]. Moreover, there is evidence that CHD patients have increased levels of asialylated LDLs characterized by the absence of terminal sialic acids on the glycans of protein chains[154]. LDL changes its density, particle size, lipid composition, and charge after desialylation[20]. DesLDL is subjected to another atherogenic modification of LDL, such as oxidation[20]. Consequently, LDL is considered multi-modified LDL, characterized by reduced sialic acid content, changes in lipid composition, reduced particle size, and acquisition of a negative charge[13,20,155]. There is an assumption that the desLDL subfraction may be electronegative LDL[121].
Furthermore, desLDL tends to aggregate, increasing its atherogenicity[12,156]. Active uptake of multi-modified LDL by arterial cells has been demonstrated by the addition of desLDL to aortic intima SMCs[157]. This phenomenon has been confirmed in experiments on mouse models. Injection of fluorescence-labeled desLDL to Apoe-/- mice resulted in active lipoprotein uptake by arterial cells[143]. In addition, desLDL can cause intracellular lipid accumulation[158]. This finding was confirmed in a study using cultured human aortic intima cells due to the increased uptake and low rate of intracellular degradation of desLDL. It has also been shown that uptake of desLDL by macrophages can be mediated by a lectin receptor such as asialoglycoprotein receptor 1[143,159]. Increased proliferative activity and the synthesis of fibrous extracellular matrix components were found during intracellular lipid accumulation induced by desLDL[160]. These events are the first evidence of cellular aspects of atherosclerosis.
The role of LDL desialylation in atherogenesis is undeniable; nevertheless, further study of the nature of sialidase activity in humans is required. Further studies will allow the modeling of LDL desialylation in animals.
GLYCATED LDL
Mechanism of glycation and effects of gLDL
Glycation is an atherogenic LDL modification. The cause of gLDL production is glycation, i.e., non-enzymatic glycosylation in hyperglycemia[161]. Non-enzymatic glycosylation increases the total negative charge of modified LDL[162]. The carbonyl groups of glucose interact with the free amino groups of ApoB-100, resulting in the formation of a Schiff base, which is converted to Amadori products via rearrangement, which in turn are converted to advanced glycation end products (AGEs) [Figure 5]. The glycation process is revealed in patients with diabetes mellitus and metabolic syndrome[163]. Incubation of native LDL with glucose resulted in dose-dependent glycation of LDL and increased LPO in vitro[164]. Glycation of the free amino groups of ApoB-100 lysine occurs in the LDLR binding domain. This event leads to the decreased affinity of LDL to their receptor and an increased average plasma lifetime[165,166]; gLDL binds to human macrophage scavenger receptors, which are hypothesized to promote the intracellular accumulation of CE and foam cell formation in atherosclerotic lesions[17].
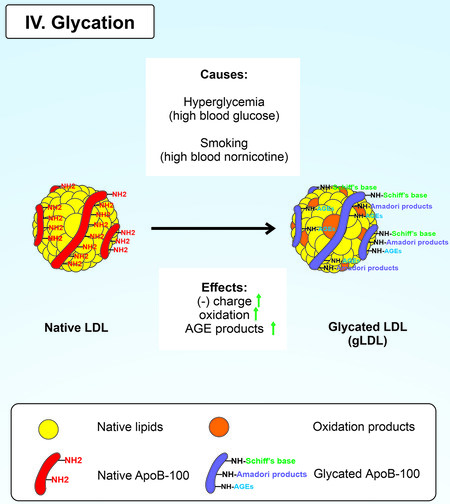
Figure 5. Glycation of native LDL.
The high degree of LDL glycation contributes to LDL oxidation, leading to the formation of highly oxidized LDL and increasing LDL atherogenicity[19,167]. Particles such as small dense LDL are most susceptible to glycation, even in non-diabetic patients[164,168,169]. A subfraction of in vivo modified atherogenic LDL was found in the blood of patients with diabetes mellitus[17]; this was a subfraction of small dense electronegative desialylated and glycated LDL that induced intracellular lipid accumulation in cell culture. It is critical to note that the synergistic effect of several LDL modifications (particularly glycation and desialylation) on the enhancement of intracellular lipid accumulation has been demonstrated in an experiment with cultured aortic SMCs obtained from healthy donors[170,171].
Non-enzymatic glycation also produces AGEs from the interaction of aldehyde groups of the reducing sugars that interact with proteins, lipids, and nucleic acids[172]. In addition to diabetes, another risk factor for CVD and myocardial infarction is smoking[173-175]. Elevated levels of ApoB-100 and albumin AGEs have been found in the blood of smokers[176]. The nicotine metabolite nornicotine explains this in the blood, which causes aberrant protein glycation[177]. The process of formation of AGE-protein adducts is irreversible. AGE-protein adducts are characterized by high stability[178]. In addition, the interaction of AGE with its receptor leads to increased expression, oxidative stress, and the release of pro-inflammatory agents[179]. AGE formation induces atherosclerosis through the proliferation of vascular SMCs, increased expression of pro-atherogenic mediators, and vascular remodeling[180].
These findings suggest the critical role of classical risk factors for atherosclerosis, such as smoking and diabetes mellitus[181-183].
CARBAMYLATED LDL
Mechanism of LDL carbamylation
The final post-translational modification of LDL is carbamylation. The carbamoyl moiety of cyanate or thiocyanate non-enzymatically binds to the free functional amino groups of ApoB-100 LDL to form carbamylated LDL (cLDL) [Figure 6][184]. This modification changes proteins’ structural and functional properties, causing partial or complete loss of functionality, disruption of protein-protein interactions, and binding to receptors[185]. Carbamylation of LDL increases the particle's electrophoretic mobility because the modified lysine’s positive charge is neutralized[186].
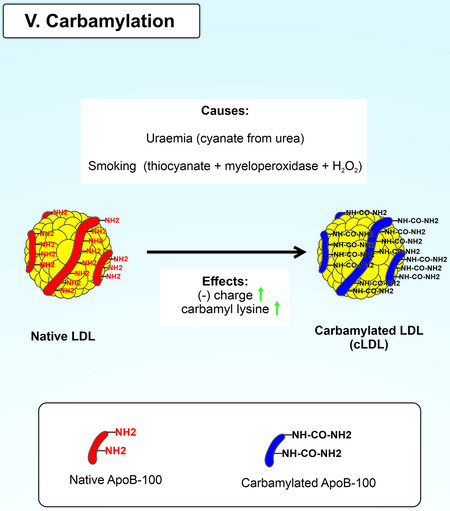
Figure 6. Carbamylation of native LDL.
Patients with chronic kidney disease (CKD) and smokers were found to have an increased risk of developing atherosclerosis[187,188]. The enhanced process of carbamylation explains these patterns in patients due to increased concentrations of urea and thiocyanate. Active urea breakdown results in elevated blood cyanate levels, followed by carbamylation of proteins in CKD patients[189]. This finding was confirmed in a study using oral administration of urea to Apoe-/-mice leading to an eight-fold increase in blood cLDL levels and more severe progression of atherosclerosis than in control mice[190]. Due to myeloperoxidase, there is an alternative mechanism in which thiocyanate is oxidized in the presence of H2O2 to cyanate in smokers[191,192].
Effects of carbamylated LDL
The atherogenic properties of cLDL are well-known; cLDL is prone to oxidation and has high cytotoxicity for endothelial cells in vitro[18]. Elevated levels of the soluble form of the lectin-like oxidized low-density lipoprotein receptor-1 and cLDL significantly increase the risk of CHD in patients with metabolic syndrome[193]. Moreover, cLDL activates the LOX-1 receptor, promoting a prothrombotic effect in vascular cells and platelets in mice[194]. Endothelial cell injury occurs due to the induction of autophagy proteins such as LC3-I, beclin-1, and Atg5 in response to cLDL[195]. In another study, cLDL promoted monocyte recruitment, adhesion to endothelial cells, and the proliferation of SMCs in coronary arteries[196,197]. The atherogenic effect of cLDL has been demonstrated in Apoe-/-mice with surgically-induced CKD. Uremic mice with high cLDL levels had more atherosclerotic lesions than control animals[190]. In addition, cLDL has reduced clearance from the blood, which may also lead to atherosclerosis progression[198].
There is robust evidence linking elevated cLDL levels to progressive atherosclerosis in humans and animal models. Nevertheless, the direct role of carbamylated LDL in the mechanism of atherogenesis remains to be established.
CONCLUSION
Native LDL is subjected to many modifications in pathological processes that increase lipoprotein atherogenicity. It is most likely that several types of LDL modifications are involved in atherogenesis. Further studies of atherogenesis in animal models will allow the use of modified LDL as a biomarker for diagnosing the subclinical form of atherosclerosis in humans. Likely, promising therapeutic strategies affecting atherogenesis will effectively treat atherosclerosis. Inhibitors of sialidases, sphingomyelinase, phospholipase A2, specific antioxidants, and substances that prevent the aggregation and uptake of modified LDL by cells can be used as probable therapeutic approaches.
It is also critical to focus on the study of risk factors for atherosclerosis, including hypertension, diabetes mellitus, smoking, and obesity, which contribute to atherogenic modifications of native LDL. Studies of the association of risk factors with atherogenesis will reveal reliable biomarkers and improve the prevention of CVDs.
DECLARATIONS
Authors’ contributionsConceptualized the manuscript: Kashirskikh DA, Guo S
Wrote the manuscript text and made a visualization: Kashirskikh DA
Reviewed the text: Bagheri Ekta M, Guo S, Grechko AV, Bogatyreva AI
Developed the methodology: Bagheri Ekta M, Chicherina NR
Completed the formal analysis: Panyod S, Guo S
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the Russian Science Foundation (Grant #22-25-00391).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Gatto L, Prati F. Subclinical atherosclerosis: how and when to treat it? Eur Hear J Suppl 2020;22:E87-90.
2. Borén J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol 2016;27:473-83.
3. Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol 1995;15:551.
4. Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res 2009;50:S376-81.
5. Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol 2006;6:508-19.
6. Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res 2016;118:620-36.
7. Orekhov AN, Myasoedova VA. Low density lipoprotein-induced lipid accumulation is a key phenomenon of atherogenesis at the arterial cell level. Vessel Plus 2019;3:3.
8. Mezentsev A, Bezsonov E, Kashirskikh D, et al. Proatherogenic sialidases and desialylated lipoproteins: 35 years of research and current state from bench to bedside. Biomedicines 2021;9:600.
9. Orekhov AN, Tertov VV, Mukhin DN, et al. Modification of low density lipoprotein by desialylation causes lipid accumulation in cultured cells: discovery of desialylated lipoprotein with altered cellular metabolism in the blood of atherosclerotic patients. Biochem Biophys Res Commun 1989;162:206-11.
10. Orsó E, Grandl M, Schmitz G. Oxidized LDL-induced endolysosomal phospholipidosis and enzymatically modified LDL-induced foam cell formation determine specific lipid species modulation in human macrophages. Chem Phys Lipids 2011;164:479-87.
11. Zhang CP, Ding XX, Tian T, et al. Impaired lipophagy in endothelial cells with prolonged exposure to oxidized low-density lipoprotein. Mol Med Rep 2020;22:2665.
12. Jauhiainen M, Ehnholm C, Gabbasov ZA, et al. Three types of naturally occurring modified lipoproteins induce intracellular lipid accumulation in human aortic intimal cells - the role of lipoprotein aggregation. Clin Chem Lab Med 1992;30:171-8.
13. Zakiev ER, Sobenin IA, Sukhorukov VN, et al. Carbohydrate composition of circulating multiple-modified low-density lipoprotein. Vasc Health Risk Manag 2016;12:379.
14. Malle E, Waeg G, Schreiber R, et al. Immunohistochemical evidence for the myeloperoxidase/H2O2/halide system in human atherosclerotic lesions: colocalization of myeloperoxidase and hypochlorite-modified proteins. Eur J Biochem 2000;267:4495-503.
15. Delporte C, Van Antwerpen P, Vanhamme L, et al. Low-density lipoprotein modified by myeloperoxidase in inflammatory pathways and clinical studies. Mediators Inflamm 2013;2013:971579.
16. Chellan B, Rojas E, Zhang C, et al. Enzyme-modified non-oxidized LDL (ELDL) induces human coronary artery smooth muscle cell transformation to a migratory and osteoblast-like phenotype. Sci Rep 2018;8:1-14.
18. Apostolov EO, Ok E, Burns S, et al. Carbamylated-oxidized LDL: proatherosclerotic effects on endothelial cells and macrophages. J Atheroscler Thromb 2013;20:878.
19. Ravandi A, Kuksis A, Shaikh NA. Glucosylated glycerophosphoethanolamines are the major LDL glycation products and increase LDL susceptibility to oxidation: evidence of their presence in atherosclerotic lesions. Arterioscler Thromb Vasc Biol 2000;20:467-77.
20. Alipov VI, Sukhorukov VN, Karagodin VP, et al. Chemical composition of circulating native and desialylated low density lipoprotein: what is the difference? Vessel Plus 2017;1:107-15.
21. Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem 1983;52:223-61.
22. Rudenko G, Henry L, Henderson K, et al. Structure of the LDL receptor extracellular domain at endosomal pH. Science 2002;298:2353-8.
23. Goldstein JL, Ho YK, Basu SK, et al. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci USA 1979;76:333.
24. Kodama T, Reddy P, Kishimoto C, et al. Purification and characterization of a bovine acetyl low density lipoprotein receptor. Proc Natl Acad Sci USA 1988;85:9238.
25. Steinbrecher UP, Parthasarathy S, Leake DS, et al. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc Natl Acad Sci USA 1984;81:3883-7.
26. Morel DW, Hessler JR, Chisolm GM. Low density lipoprotein cytotoxicity induced by free radical peroxidation of lipid. J Lipid Res 1983;24:1070-6.
27. Friedman P, Hörkkö S, Steinberg D, et al. Correlation of antiphospholipid antibody recognition with the structure of synthetic oxidized phospholipids. Importance of Schiff base formation and aldol condensation. J Biol Chem 2002;277:7010-20.
28. Yia-Herttuala S, Palinski W, Rosenfeld ME, et al. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Investig 1989;84:1086.
29. Burtenshaw D, Kitching M, Redmond EM, et al. Reactive oxygen species (ROS), intimal thickening, and subclinical atherosclerotic disease. Front Cardiovasc Med 2019;6:89.
30. Gao S, Zhao D, Qi Y, et al. Circulating oxidized low-density lipoprotein levels independently predict 10-year progression of subclinical carotid atherosclerosis: a community-based cohort study. J Atheroscler Thromb 2018;25:1032.
31. Anggraeni VY, Emoto N, Yagi K, et al. Correlation of C4ST-1 and ChGn-2 expression with chondroitin sulfate chain elongation in atherosclerosis. Biochem Biophys Res Commun 2011;406:36-41.
32. Williams KJ. Arterial wall chondroitin sulfate proteoglycans: diverse molecules with distinct roles in lipoprotein retention and atherogenesis. Curr Opin Lipidol 2001;12:477-87.
33. Kaplan M, Aviram M. Macrophage plasma membrane chondroitin sulfate proteoglycan binds oxidized low-density lipoprotein. Atherosclerosis 2000;149:5-17.
34. Adhikara IM, Yagi K, Mayasari DS, et al. Chondroitin sulfate N-acetylgalactosaminyltransferase-2 impacts foam cell formation and atherosclerosis by altering macrophage glycosaminoglycan chain. Arterioscler Thromb Vasc Biol 2021;41:1076-91.
35. Steinberg D, Witztum JL. History of discovery: oxidized low-density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol 2010;30:2311-6.
36. Tertov VV, Sobenin IA, Kaplun VV, et al. Antioxidant content in low density lipoprotein and lipoprotein oxidation in vivo and in vitro. Free Radic Res 1998;29:165-73.
37. Rhoads JP, Major AS. How oxidized low-density lipoprotein activates inflammatory responses. Crit Rev Immunol 2018;38:333-42.
38. Maiolino G, Rossitto G, Caielli P, et al. The role of oxidized low-density lipoproteins in atherosclerosis: the myths and the facts. Mediators Inflamm 2013;2013:714653.
39. Steinberg D. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat Med 2002;8:1211-7.
40. Wen Y, Leake DS. Low density lipoprotein undergoes oxidation within lysosomes in cells. Circ Res 2007;100:1337-43.
41. Ahmad F, Leake DS. Antioxidants inhibit low density lipoprotein oxidation less at lysosomal pH: a possible explanation as to why the clinical trials of antioxidants might have failed. Chem Phys Lipids 2018;213:13.
42. Ahmad F, Leake DS. Lysosomal oxidation of LDL alters lysosomal pH, induces senescence, and increases secretion of pro-inflammatory cytokines in human macrophages. J Lipid Res 2019;60:98.
43. Kattoor AJ, Pothineni NVK, Palagiri D, et al. Oxidative stress in atherosclerosis. Curr Atheroscler Rep 2017;19:42.
44. Ke C, Zhu X, Zhang Y, et al. Metabolomic characterization of hypertension and dyslipidemia. Metabolomics 2018;14:117.
45. Burleigh ME, Babaev VR, Oates JA, et al. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in LDL receptor-deficient mice. Circulation 2002;105:1816-23.
46. Belkner J, Chaitidis P, Stender H, et al. Expression of 12/15-lipoxygenase attenuates intracellular lipid deposition during in vitro foam cell formation. Arterioscler Thromb Vasc Biol 2005;25:797-802.
47. Carr AC, McCall MR, Frei B. Oxidation of LDL by myeloperoxidase and reactive nitrogen species. Arterioscler Thromb Vasc Biol 2000;20:1716-23.
48. Dieber-Rotheneder M, Waeg G, Striegl G, et al. Biochemical, structural, and functional properties of oxidized low-density lipoprotein. Chem Res Toxicol 1990;3:77-92.
49. Ito J, Shimizu N, Kato S, et al. Direct separation of the diastereomers of cholesterol ester hydroperoxide using LC-MS/MS to evaluate enzymatic lipid oxidation. Symmetry 2020;12:1127.
50. Piotrowski JJ, Shah S, Alexander JJ. Mature human atherosclerotic plaque contains peroxidized phosphatidylcholine as a major lipid peroxide. Life Sci 1996;58:735-40.
51. Niki E, Noguchi N. Dynamics of oxidation of LDL and its inhibition by antioxidants. BioFactors 1997;6:201-8.
52. Belkner J, Stender H, Kühn H. The rabbit 15-lipoxygenase preferentially oxygenates LDL cholesterol esters, and this reaction does not require vitamin E. J Biol Chem 1998;273:23225-32.
53. Takahashi Y, Glasgow WC, Suzuki H, et al. Investigation of the oxygenation of phospholipids by the porcine leukocyte and human platelet arachidonate 12-lipoxygenases. Eur J Biochem 1993;218:165-71.
54. Yamamoto S. Mammalian lipoxygenases: molecular structures and functions. Biochim Biophys Acta 1992;1128:117-31.
55. Funk CD. The molecular biology of mammalian lipoxygenases and the quest for eicosanoid functions using lipoxygenase-deficient mice. Biochim Biophys Acta 1996;1304:65-84.
56. Zhu H, Takahashi Y, Xu W, et al. Low density lipoprotein receptor-related protein-mediated membrane translocation of 12/15-lipoxygenase is required for oxidation of low density lipoprotein by macrophages. J Biol Chem 2003;278:13350-5.
57. Takahashi Y, Zhu H, Xu W, et al. Selective uptake and efflux of cholesteryl linoleate in LDL by macrophages expressing 12/15-lipoxygenase. Biochem Biophys Res Commun 2005;338:128-35.
58. Singh NK, Rao GN. Emerging role of 12/15-lipoxygenase (ALOX15) in human pathologies. Prog Lipid Res 2019;73:28.
59. Belkner J, Wiesner R, Rathman J, et al. Oxygenation of lipoproteins by mammalian lipoxygenases. Eur J Biochem 1993;213:251-61.
60. Parthasarathy S, Wieland E, Steinberg D. A role for endothelial cell lipoxygenase in the oxidative modification of low density lipoprotein. Proc Natl Acad Sci USA 1989;86:1046-50.
61. Harats D, Shaish A, George J, et al. Overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol 2000;20:2100-5.
62. Ylä-Herttuala S, Rosenfeld ME, Parthasarathy S, et al. Colocalization of 15-lipoxygenase mRNA and protein with epitopes of oxidized low density lipoprotein in macrophage-rich areas of atherosclerotic lesions. Proc Natl Acad Sci USA 1990;87:6959.
63. Hiltunen T, Luoma J, Nikkari T, et al. Induction of 15-lipoxygenase mRNA and protein in early atherosclerotic lesions. Circulation 1995;92:3297-303.
64. Santos LRB, Fleming I. Role of cytochrome P450-derived, polyunsaturated fatty acid mediators in diabetes and the metabolic syndrome. Prostaglandins Other Lipid Mediat 2020;148:106407.
65. Gomez I, Foudi N, Longrois D, et al. The role of prostaglandin E2 in human vascular inflammation. Prostaglandins Leukot Essent Fatty Acids 2013;89:55-63.
66. Tang SY, Monslow J, Todd L, et al. Cyclooxygenase-2 in endothelial and vascular smooth muscle cells restrains atherogenesis in hyperlipidemic mice. Circulation 2014;129:1761.
67. Spector AA, Kim HY. Cytochrome P450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim Biophys Acta 2015;1851:356.
68. Fissithaler B, Popp R, Kiss L, et al. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 1999;401:493-7.
69. Node K, Huo Y, Ruan X, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999;285:1276-9.
70. Campbell WB, Gebremedhin D, Pratt PF, et al. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res 1996;78:415-23.
72. Baldus S, Heeschen C, Meinertz T, et al. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation 2003;108:1440-5.
73. Brennan ML, Penn MS, Van Lente F, et al. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med 2003;349:1595-604.
74. Halliwell B, Gutteridge JMC. Oxygen free radicals and iron in relation to biology and medicine: some problems and concepts. Arch Biochem Biophys 1986;246:501-14.
75. Lynch SM, Frei B. Mechanisms of copper- and iron-dependent oxidative modification of human low density lipoprotein. J Lipid Res 1993;3:1745-53.
76. Zhang Y, Murugesan P, Huang K, et al. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat Rev Cardiol 2020;17:170.
77. Nakajima K, Nakano T, Tanaka A. The oxidative modification hypothesis of atherosclerosis: the comparison of atherogenic effects on oxidized LDL and remnant lipoproteins in plasma. Clin Chim Acta 2006;367:36-47.
78. Cheng C, Zheng E, Yu B, et al. Recognition of lipoproteins by scavenger receptor class A members. J Biol Chem 2021;297:100948.
79. Kunjathoor VV, Febbraio M, Podrez EA, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem 2002;277:49982-8.
80. Chen B, Meng L, Shen T, et al. Thioredoxin attenuates oxidized low-density lipoprotein induced oxidative stress in human umbilical vein endothelial cells by reducing NADPH oxidase activity. Biochem Biophys Res Commun 2017;490:1326-33.
81. Frostegard J, Nilsson J, Haegerstrand A, et al. Oxidized low density lipoprotein induces differentiation and adhesion of human monocytes and the monocytic cell line U937. Proc Natl Acad Sci USA 1990;87:904.
82. Hansson GK. Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol 2001;21:1876-90.
83. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature 2011;473:317-25.
84. Li Y, Li Y, Yang T, et al. Dioscin attenuates oxLDL uptake and the inflammatory reaction of dendritic cells under high glucose conditions by blocking p38 MAPK. Mol Med Rep 2020;21:304-10.
85. Gorog P, Born GVR. Nifedipine inhibits accumulation of LDL and cholesterol in the aorta of the normocholesterolemic rabbit. Arterioscler Thromb Vasc Biol 1993;13:637-9.
86. Bhakdi S, Dorweiler B, Kirchmann R, et al. On the pathogenesis of atherosclerosis: enzymatic transformation of human low density lipoprotein to an atherogenic moiety. J Exp Med 1995;182:1959-71.
87. Maaninka K, Nguyen SD, Mäyränpää MI, et al. Human mast cell neutral proteases generate modified LDL particles with increased proteoglycan binding. Atherosclerosis 2018;275:390-9.
88. Torzewski M, Suriyaphol P, Paprotka K, et al. Enzymatic modification of low-density lipoprotein in the arterial wall: a new role for plasmin and matrix metalloproteinases in atherogenesis. Arterioscler Thromb Vasc Biol 2004;24:2130-6.
89. Han SR, Momeni A, Strach K, et al. Enzymatically modified LDL induces cathepsin H in human monocytes: potential relevance in early atherogenesis. Arterioscler Thromb Vasc Biol 2003;23:661-7.
90. Öörni K, Sneck M, Brömme D, et al. Cysteine protease cathepsin F is expressed in human atherosclerotic lesions, is secreted by cultured macrophages, and modifies low density lipoprotein particles in vitro. J Biol Chem 2004;279:34776-84.
91. Lindstedt L, Lee M, Castro GR, et al. Chymase-containing mast cells in human arterial intima: implications for atherosclerotic disease. J Clin Investig 1996;97:2174-82.
92. Öörni K, Hakala JK, Annila A, et al. Sphingomyelinase induces aggregation and fusion, but phospholipase A2 only aggregation, of low density lipoprotein (LDL) particles. Two distinct mechanisms leading to increased binding strength of LDL to human aortic proteoglycans. J Biol Chem 1998;273:29127-34.
93. Piha M, Lindstedt L, Kovanen PT. Fusion of proteolyzed low-density lipoprotein in the fluid phase: a novel mechanism generating atherogenic lipoprotein particles. Biochemistry 1995;34:10120-9.
94. Chellan B, Reardon CA, Getz GS, et al. Enzymatically modified low-density lipoprotein promotes foam cell formation in smooth muscle cells via macropinocytosis and enhances receptor-mediated uptake of oxidized low-density lipoprotein. Arterioscler Thromb Vasc Biol 2016;36:1101-13.
95. Hui DY. Phospholipase A2 enzymes in metabolic and cardiovascular diseases. Curr Opin Lipidol 2012;23:235.
96. Elinder LS, Dumitrescu A, Larsson P, et al. Expression of phospholipase A2 isoforms in human normal and atherosclerotic arterial wall. Arterioscler Thromb Vasc Biol 1997;17:2257-63.
97. Kolodgie FD, Burke AP, Skorija KS, et al. Lipoprotein-associated phospholipase A2 protein expression in the natural progression of human coronary atherosclerosis. Arterioscler Thromb Vasc Biol 2006;26:2523-9.
98. Lehti S, Nguyen SD, Belevich I, et al. Extracellular lipids accumulate in human carotid arteries as distinct three-dimensional structures and have proinflammatory properties. Am J Pathol 2018;188:525-38.
99. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010;464:1357-61.
100. Stary HC, Chandler AB, Dinsmore RE, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis, American Heart Association. Circulation 1995;92:1355-74.
101. Hakala JK, Oksjoki R, Laine P, et al. Lysosomal enzymes are released from cultured human macrophages, hydrolyze LDL in vitro, and are present extracellularly in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol 2003;23:1430-6.
102. Heffron SP, Ruuth MK, Xia Y, et al. Low-density lipoprotein aggregation predicts adverse cardiovascular events in peripheral artery disease. Atherosclerosis 2021;316:53-7.
103. Ruuth M, Nguyen SD, Vihervaara T, et al. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur Heart J 2018;39:2562-73.
104. Torzewski M. Enzymatically modified LDL, atherosclerosis and beyond: paving the way to acceptance. Front Biosci 2018;23:1257-71.
105. Arlaud GJ, Biro A, Ling WL. Enzymatically modified low-density lipoprotein is recognized by c1q and activates the classical complement pathway. J Lipids 2011;2011:1-5.
106. Martin-Ventura JL, Martinez-Lopez D, Roldan-Montero R, et al. Role of complement system in pathological remodeling of the vascular wall. Mol Immunol 2019;114:207-15.
107. Oksjoki R, Kovanen PT, Pentikäinen MO. Role of complement activation in atherosclerosis. Curr Opin Lipidol 2003;14:477-82.
108. Oestvang J, Bonnefont-Rousselot D, Ninio E, et al. Modification of LDL with human secretory phospholipase A2 or sphingomyelinase promotes its arachidonic acid-releasing propensity. J Lipid Res 2004;45:831-8.
109. Taylor J, Butcher M, Zeadin M, et al. Oxidized low-density lipoprotein promotes osteoblast differentiation in primary cultures of vascular smooth muscle cells by up-regulating Osterix expression in an Msx2-dependent manner. J Cell Biochem 2011;112:581-8.
110. Liu J, Ren Y, Kang L, et al. Oxidized low-density lipoprotein increases the proliferation and migration of human coronary artery smooth muscle cells through the upregulation of osteopontin. Int J Mol Med 2014;33:1341-7.
111. Newby AC. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc Med 2007;17:253-8.
112. Myasoedova VA, Di Minno A, Songia P, et al. Sex-specific differences in age-related aortic valve calcium load: a systematic review and meta-analysis. Ageing Res Rev 2020;61:101077.
113. Twardowski L, Cheng F, Michaelsen J, et al. Enzymatically modified low-density lipoprotein is present in all stages of aortic valve sclerosis: implications for pathogenesis of the disease. J Am Heart Assoc 2015;4:e002156.
114. Pavoine C, Pecker F. Sphingomyelinases: their regulation and roles in cardiovascular pathophysiology. Cardiovasc Res 2009;82:175.
115. Wang W, Zhang J, Wu W, et al. Inhibition of lipoprotein-associated phospholipase A2 ameliorates inflammation and decreases atherosclerotic plaque formation in ApoE-deficient mice. PLoS One 2011;6:e23425.
116. Melnichenko AA, Aksenov DV, Myasoedova VA, et al. Pluronic block copolymers inhibit low density lipoprotein self-association. Lipids 2012;47:995-1000.
117. Chazov EI, Orekhov AN, Perova NV, et al. Atherogenicity of blood serum from patients with coronary heart disease. Lancet 1986;328:595-8.
118. Orekhov AN, Tertov VV, Pokrovsky SN, et al. Blood serum atherogenicity associated with coronary atherosclerosis. Evidence for nonlipid factor providing atherogenicity of low-density lipoproteins and an approach to its elimination. Circ Res 1988;62:421-9.
119. Tertov VV, Orekhov AN, Kacharava AG, et al. Low density lipoprotein-containing circulating immune complexes and coronary atherosclerosis. Exp Mol Pathol 1990;52:300-8.
120. Tertov VV, Sobenin IA, Tonevitsky AG, et al. Isolation of atherogenic modified (desialylated) low density lipoprotein from blood of atherosclerotic patients: separation from native lipoprotein by affinity chromatography. Biochem Biophys Res Commun 1990;167:1122-7.
121. Tertov VV, Bittolo-Bon G, Sobenin IA, et al. Naturally occurring modified low density lipoproteins are similar if not identical: more electronegative and desialylated lipoprotein subfractions. Exp Mol Pathol 1995;62:166-72.
122. Tertov VV, Sobenin IA, Gabbasov ZA, et al. Multiple-modified desialylated low density lipoproteins that cause intracellular lipid accumulation. Isolation, fractionation and characterization. Lab Invest 1992;67:665-75.
123. Tertov VV, Kaplun VV, Sobenin IA, et al. Human plasma trans-sialidase causes atherogenic modification of low density lipoprotein. Atherosclerosis 2001;159:103-15.
124. Glanz VY, Myasoedova VA, Grechko AV, et al. Trans-sialidase associated with atherosclerosis: defining the identity of a key enzyme involved in the pathology. Curr Drug Targets 2019;20:938-41.
125. Dobie C, Skropeta D. Insights into the role of sialylation in cancer progression and metastasis. Br J Cancer 2021;124:76.
126. Li F, Ding J. Sialylation is involved in cell fate decision during development, reprogramming and cancer progression. Protein Cell 2019;10:550.
127. MacAuley MS, Crocker PR, Paulson JC. Siglec regulation of immune cell function in disease. Nat Rev Immunol 2014;14:653.
128. Zhang C, Chen J, Liu Y, et al. Sialic acid metabolism as a potential therapeutic target of atherosclerosis. Lipids Health Dis 2019;18:173.
129. Makarava N, Chang JCY, Baskakov IV. Region-specific sialylation pattern of prion strains provides novel insight into prion neurotropism. Int J Mol Sci 2020;21:828.
130. Bongiovanni A, Cusimano A, Annunziata I, et al. Sialylation of host proteins as targetable risk factor for COVID-19 susceptibility and spreading: a hypothesis. FASEB Bioadv 2021;3:192.
131. Betteridge KB, Arkill KP, Neal CR, et al. Sialic acids regulate microvessel permeability, revealed by novel in vivo studies of endothelial glycocalyx structure and function. J Physiol 2017;595:5015.
132. Eguchi H, Ikeda Y, Ookawara T, et al. Modification of oligosaccharides by reactive oxygen species decreases sialyl lewis x-mediated cell adhesion. Glycobiology 2005;15:1094-101.
133. Tanaka K, Tokumaru S, Kojo S. Possible involvement of radical reactions in desialylation of LDL. FEBS Lett 1997;413:202-4.
134. Iijima R, Takahashi H, Ikegami S, et al. Characterization of the reaction between sialic acid (N-acetylneuraminic acid) and hydrogen peroxide. Biol Pharm Bull 2007;30:580-2.
135. Yasuda J, Eguchi H, Fujiwara N, et al. Reactive oxygen species modify oligosaccharides of glycoproteins in vivo: a study of a spontaneous acute hepatitis model rat (LEC rat). Biochem Biophys Res Commun 2006;342:127-34.
136. Öztürk Z, Sönmez H, Görgün FM, et al. The relationship between lipid peroxidation and LDL desialylation in experimental atherosclerosis. Toxicol Mech Methods 2007;17:265-73.
137. Sukhorukov V, Gudelj I, Pučić-Baković M, et al. Glycosylation of human plasma lipoproteins reveals a high level of diversity, which directly impacts their functional properties. Biochim Biophys Acta Mol Cell Biol Lipids 2019;1864:643-53.
138. Igdoura SA, Gafuik C, Mertineit C, et al. Cloning of the cDNA and gene encoding mouse lysosomal sialidase and correction of sialidase deficiency in human sialidosis and mouse SM/J fibroblasts. Hum Mol Genet 1998;7:115-20.
139. Monti E, Preti A, Rossi E, et al. Cloning and characterization of NEU2, a human gene homologous to rodent soluble sialidases. Genomics 1999;57:137-43.
140. Miyagi T, Wada T, Iwamatsu A, et al. Molecular cloning and characterization of a plasma membrane-associated sialidase specific for gangliosides. J Biol Chem 1999;274:5004-11.
141. Comelli EM, Amado M, Lustig SR, et al. Identification and expression of Neu4, a novel murine sialidase. Gene 2003;321:155-61.
142. White EJ, Gyulay G, Lhoták Š, et al. Sialidase down-regulation reduces non-HDL cholesterol, inhibits leukocyte transmigration, and attenuates atherosclerosis in ApoE knockout mice. J Biol Chem 2018;293:14689.
143. Demina EP, Smutova V, Pan X, et al. Neuraminidases 1 and 3 trigger atherosclerosis by desialylating low-density lipoproteins and increasing their uptake by macrophages. J Am Hear Assoc Cardiovasc Cerebrovasc Dis 2021;10:1-38.
144. Bowles WHD, Gloster TM. Sialidase and sialyltransferase inhibitors: targeting pathogenicity and disease. Front Mol Biosci 2021;8:705.
145. Tertov VV, Nikonova EY, Nifant’ev NE, et al. Human plasma trans-sialidase donor and acceptor specificity. Biochemistry 2002;67:908-13.
146. Mehr K, Withers SG. Mechanisms of the sialidase and trans-sialidase activities of bacterial sialyltransferases from glycosyltransferase family 80. Glycobiology 2016;26:353-9.
148. Suo J, Zhao L, Wang J, et al. Influenza virus aggravates the ox-LDL-induced apoptosis of human endothelial cells via promoting p53 signaling. J Med Virol 2015;87:1113-23.
149. Glanz VY, Kashirskikh DA, Grechko AV, et al. Sialidase activity in human blood serum has a distinct seasonal pattern: a pilot study. Biology 2020;9:1-7.
150. Cha SK, Ortega B, Kurosu H, et al. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci USA 2008;105:9805-10.
151. Cha SK, Hu MC, Kurosu H, et al. Regulation of renal outer medullary potassium channel and renal K+ excretion by Klotho. Mol Pharmacol 2009;76:38-46.
152. Yao Y, Wang Y, Zhang Y, et al. Klotho ameliorates oxidized low density lipoprotein (ox-LDL)-induced oxidative stress via regulating LOX-1 and PI3K/Akt/eNOS pathways. Lipids Health Dis 2017:16.
153. Tomin A, Dumych T, Tolstyak Y, et al. Desialylation of dying cells with catalytically active antibodies possessing sialidase activity facilitate their clearance by human macrophages. Clin Exp Immunol 2015;179:17.
154. Mcdowell A, Young IS, Wisdom GB. Measurement of asialylated LDL in the blood of patients with coronary artery disease by antibody-lectin sandwich assay. Ann Clin Biochem 2001;38:499-508.
155. Sobenin IA, Galitsyna EV, Grechko AV, et al. Small dense and desialylated low density lipoprotein in diabetic patients. Vessel Plus 2017;1:29-37.
156. Tertov VV, Sobenin IA, Gabbasov ZA, et al. Lipoprotein aggregation as an essential condition of intracellular lipid accumulation caused by modified low density lipoproteins. Biochem Biophys Res Commun 1989;163:489-94.
157. Tertov VV, Orekhov AN. Metabolism of native and naturally occurring multiple modified low density lipoprotein in smooth muscle cells of human aortic intima. Exp Mol Pathol 1997;64:127-45.
158. Aksenov DV, Medvedeva LA, Skalbe TA, et al. Deglycosylation of apo B-containing lipoproteins increase their ability to aggregate and to promote intracellular cholesterol accumulation in vitro. Arch Physiol Biochem 2008;114:349-56.[DOI:10.1080/13813450802227915].
159. Grewal T, Bartlett A, Burgess JW, et al. Desialylated LDL uptake in human and mouse macrophages can be mediated by a lectin receptor. Atherosclerosis 1996;121:151-63.
160. Tertov VV, Orekhov AN, Ryong LH, et al. Intracellular cholesterol accumulation is accompanied by enhanced proliferative activity of human aortic intimal cells. Tissue Cell 1988;20:849-54.
161. Doucet C, Huby T, Ruiz J, et al. Non-enzymatic glycation of lipoprotein(a) in vitro and in vivo. Atherosclerosis 1995;118:135-43.
162. Creriche AG, Stahl AJC. Glycation and oxidation of human low density lipoproteins reduces heparin binding and modifies charge. Scand J Clin Lab Invest 2009;53:125-32.
163. Akanji AO, Abdella N, Mojiminiyi OA. Determinants of glycated LDL levels in nondiabetic and diabetic hyperlipidaemic patients in Kuwait. Clin Chim Acta 2002;317:171-6.
164. Younis NN, Soran H, Pemberton P, et al. Small dense LDL is more susceptible to glycation than more buoyant LDL in type 2 diabetes. Clin Sci 2013;124:343-9.
165. Bucala R, Makita Z, Vega G, et al. Modification of low density lipoprotein by advanced glycation end products contributes to the dyslipidemia of diabetes and renal insufficiency. Proc Natl Acad Sci USA 1994;91:9441.
166. Witztum JL, Mahoney EM, Branks MJ, et al. Nonenzymatic glucosylation of low-density lipoprotein alters its biologic activity. Diabetes 1982;31:283-91.
167. Siddiqui K, George TP, Nawaz SS, et al. Significance of glycated LDL in different stages of diabetic nephropathy. Diabetes Metab Syndr Clin Res Rev 2019;13:548-52.
168. Bowie A, Owens D, Collins P, et al. Glycosylated low density lipoprotein is more sensitive to oxidation: implications for the diabetic patient? Atherosclerosis 1993;102:63-7.
169. Younis N, Charlton-Menys V, Sharma R, et al. Glycation of LDL in non-diabetic people: small dense LDL is preferentially glycated both in vivo and in vitro. Atherosclerosis 2009;202:162-8.
170. Sobenin IA, Tertov VV, Koschinsky T, et al. Modified low density lipoprotein from diabetic patients causes cholesterol accumulation in human intimal aortic cells. Atherosclerosis 1993;100:41-54.
171. Sobenin IA, Tertov VV, Orekhov AN, et al. Synergetic effect of desialylated and glycated low density lipoproteins on cholesterol accumulation in cultured smooth muscle intimal cells. Atherosclerosis 1991;89:151-4.
172. Turco S, Basta G. An update on advanced glycation endproducts and atherosclerosis. BioFactors 2012;38:266-74.
173. De Michele G, Correale M, De Michele O, et al. Evaluation of serum biomarkers in nutritional disorders: glycated apolipoprotein B, fasting serum glucose, fructosamine, stable and labile glycated hemoglobin in diabetic and non-diabetic subjects. Immunopharmacol Immunotoxicol 2008;30:925-36.
174. Misciagna G, Logroscino G, De Michele G, et al. Glycated apolipoprotein B and myocardial infarction. Nutr Metab Cardiovasc Dis 2007;17:6-12.
175. Salehi N, Janjani P, Tadbiri H, et al. Effect of cigarette smoking on coronary arteries and pattern and severity of coronary artery disease: a review. J Int Med Res 2021:49.
176. Nicholl ID, Stitt AW, Moore JE, et al. Increased levels of advanced glycation endproducts in the lenses and blood vessels of cigarette smokers. Mol Med 1998;4:594.
177. Dickerson TJ, Janda KD. A previously undescribed chemical link between smoking and metabolic disease. Proc Natl Acad Sci USA 2002;99:15084.
178. Bucala R, Makita Z, Koschinsky T, et al. Lipid advanced glycosylation: pathway for lipid oxidation in vivo. Proc Natl Acad Sci USA 1993;90:6434.
179. Yuan T, Yang T, Chen H, et al. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol 2019;20:247.
180. Xiao ZL, Ma LP, Yang DF, et al. Profilin-1 is involved in macroangiopathy induced by advanced glycation end products via vascular remodeling and inflammation. World J Diabetes 2021;12:1875.
181. Mazzone A, Cusa C, Mazzucchelli I, et al. Cigarette smoking and hypertension influence nitric oxide release and plasma levels of adhesion molecules. Clin Chem Lab Med 2001;39:822-6.
182. Järvisalo MJ, Raitakari M, Toikka JO, et al. Endothelial dysfunction and increased arterial intima-media thickness in children with type 1 diabetes. Circulation 2004;109:1750-5.
183. Hajar R. Risk factors for coronary artery disease: historical perspectives. Heart Views 2017;18:109.
184. Delanghe S, Delanghe JR, Speeckaert R, et al. Mechanisms and consequences of carbamoylation. Nat Rev Nephrol 2017;13:580-93.[DOI:10.1038/NRNEPH.2017.103].
185. Jaisson S, Pietrement C, Gillery P. Protein carbamylation: chemistry, pathophysiological involvement, and biomarkers. Adv Clin Chem 2018;84:1-38.
186. Ok E, Basnakian AG, Apostolov EO, et al. Carbamylated low-density lipoprotein induces death of endothelial cells: a link to atherosclerosis in patients with kidney disease. Kidney Int 2005;68:173-8.
187. Staplin N, Haynes R, Herrington WG, et al. Smoking and adverse outcomes in patients with CKD: the study of heart and renal protection (SHARP). Am J Kidney Dis 2016;68:371.
188. Valdivielso JM, Rodríguez-Puyol D, Pascual J, et al. Atherosclerosis in chronic kidney disease. Arterioscler Thromb Vasc Biol 2019;39:1938-66.
189. Russo E, Verzola D, Cappadona F, et al. The role of uric acid in renal damage - a history of inflammatory pathways and vascular remodeling. Vessel Plus 2021;5:15.
190. Apostolov EO, Ray D, Savenka AV, et al. Chronic uremia stimulates LDL carbamylation and atherosclerosis. J Am Soc Nephrol 2010;21:1852.
191. Kumar J, Shah SV. Kidney disease as an independent risk factor for cardiovascular events. J Ren Nutr 2005;15:99-104.
192. Wang Z, Nicholls SJ, Rodriguez ER, et al. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med 2007;13:1176-84.
193. Stankova T, Delcheva G, Maneva A, et al. Serum levels of carbamylated LDL and soluble lectin-like oxidized low-density lipoprotein receptor-1 are associated with coronary artery disease in patients with metabolic syndrome. Medicina 2019;55:493.
194. Holy EW, Akhmedov A, Speer T, et al. Carbamylated low-density lipoproteins induce a prothrombotic state via LOX-1: impact on arterial thrombus formation in vivo. J Am Coll Cardiol 2016;68:1664-76.
195. Bose C, Shah SV, Karaduta OK, et al. Carbamylated low-density lipoprotein (cLDL)-mediated induction of autophagy and its role in endothelial cell injury. PLoS One 2016;11:e0165576.
196. Asci G, Basci A, Shah SV, et al. Carbamylated low-density lipoprotein induces proliferation and increases adhesion molecule expression of human coronary artery smooth muscle cells. Nephrology 2008;13:480-6.
197. Apostolov EO, Shah SV, Ok E, et al. Carbamylated low-density lipoprotein induces monocyte adhesion to endothelial cells through intercellular adhesion molecule-1 and vascular cell adhesion molecule-1. Arterioscler Thromb Vasc Biol 2007;27:826-32.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Kashirskikh DA, Guo S, Panyod S, Chicherina NR, Bagheri Ekta M, Bogatyreva AI, Grechko AV. A novel insight into the nature of modified low-density lipoproteins and their role in atherosclerosis. Vessel Plus 2023;7:3. http://dx.doi.org/10.20517/2574-1209.2022.35
AMA Style
Kashirskikh DA, Guo S, Panyod S, Chicherina NR, Bagheri Ekta M, Bogatyreva AI, Grechko AV. A novel insight into the nature of modified low-density lipoproteins and their role in atherosclerosis. Vessel Plus. 2023; 7: 3. http://dx.doi.org/10.20517/2574-1209.2022.35
Chicago/Turabian Style
Kashirskikh, Dmitry A., Shuzhen Guo, Suraphan Panyod, Nelya R. Chicherina, Mariam Bagheri Ekta, Anastasia I. Bogatyreva, Andrey V. Grechko. 2023. "A novel insight into the nature of modified low-density lipoproteins and their role in atherosclerosis" Vessel Plus. 7: 3. http://dx.doi.org/10.20517/2574-1209.2022.35
ACS Style
Kashirskikh, DA.; Guo S.; Panyod S.; Chicherina NR.; Bagheri Ekta M.; Bogatyreva AI.; Grechko AV. A novel insight into the nature of modified low-density lipoproteins and their role in atherosclerosis. Vessel Plus. 2023, 7, 3. http://dx.doi.org/10.20517/2574-1209.2022.35
About This Article
Copyright

Data & Comments
Data


 Cite This Article 26 clicks
Cite This Article 26 clicks

 Like This Article 13
likes
Like This Article 13
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.