
Features of mitochondrial dynamics in monocytes in inflammatory and metabolic disorders

Abstract
Mitochondria do not exist as separate formations in the cell; they form a homogeneous network in which the processes of division and fusion continuously occur. A shift in this balance, as well as mitochondrial dysfunction, leads to the development of chronic and metabolic disorders. Metabolic changes in mitochondria control the formation and differentiation of monocytes. Pro-inflammatory activation of monocytes/macrophages leads to a decrease in oxidative phosphorylation and an increase in mitochondrial fusion. To date, the molecular mechanisms that regulate mitochondrial dynamics to control life and death in monocytes are not well understood. In addition, there is ample evidence that abnormal mitochondrial metabolism is involved in the pathogenesis of many diseases. Mitochondrial stress and damage contribute to cell death, metabolic disorders, and inflammation. In this review, we consider in detail the involvement of mitochondrial processes in the development of pathologies and discuss how mitochondria can be therapeutically affected. Attention is also drawn to possible diagnostic studies that target mitochondrial dynamics of disorders in monocytes.
Keywords
INTRODUCTION
Chronic inflammatory diseases include metabolic disorders such as obesity and diabetes, as well as vascular diseases, including atherosclerosis[1]. The pathogenesis of these disorders is characterized by the initiation of recruited circulating monocytes into metabolic organs and tissues, as well as into the walls of blood vessels. The metabolic status of monocytes switches in response to environmental stimuli, so cellular metabolism plays a critical role in monocyte activation[2]. It is impossible to overestimate the role of mitochondria in the regulation of metabolism since they are involved in processes such as glucose oxidation and fatty acid biosynthesis[3]. The functions of mitochondria can also include the regulation of signaling pathways and differentiation of immune cells by controlling metabolic processes and the production of mitochondrial reactive oxygen species (mtROS)[4]. Mitochondrial dysfunction or disruption of mitochondrial machinery coupled with decreased mitochondrial mass limits the ability of oxidative phosphorylation to resynthesize ATP[5]. The violation of lipid metabolism leads to the accumulation of lipids in tissues. In addition, mitochondrial dysfunction affects the regulation of the production of intermediate metabolic products, reducing energy production[6]. In 2008, Kim discovered in patients with obesity and insulin resistance that changes in the content of mitochondria occur in liver tissues, adipose tissue, and skeletal muscles, and their oxidative capacity decreases[7]. Mitochondrial dysfunction is closely associated with the pathogenesis of insulin resistance and inflammation[8].
Normally, the balance of mitochondrial dynamics is maintained by their fusion and division processes, as well as mitophagy[9]. The mitochondrial fusion process involves two stages. First, the outer shells are joined, followed by the fusion of the inner shells of the mitochondria. Normally, these processes are coordinated, but mutations may appear in the genes that control them, and fusion and division become independent of each other[10].
Maintenance of repair is carried out in the process of mitochondrial division by separating the damaged organelle and degradation by mitophagy of non-functioning companions. It should be noted that the metabolic processes in the cell can be disrupted by excessive mitochondrial division[11]. Fusion and fission provide an exchange between membrane lipids and intramitochondrial contents in mitochondria, which is very important for maintaining a normal mitochondrial population[12]. Any disturbance in mitochondrial dynamics that alters the normal balance between fission and fusion can lead to an accumulation of damaged and dysfunctional organelles. Thus, mitochondrial dynamics play an important role in the morphology, function, and distribution of mitochondria.
Mitophagy is considered to be one of the main processes of quality control of mitochondrial mechanisms. This is a form of autophagy that selectively removes damaged mitochondria by means of the autophagosomal–lysosomal apparatus[13]. Mitophagy is very important to prevent oxidative damage in the cell, which can affect the enzymes of the respiratory chain.
The purpose of this review is to describe and summarize data on the mechanisms that occur in monocyte mitochondria during inflammatory and metabolic disorders.
FORMATION OF CLASSIC MONOCYTES IN NORMAL AND PATHOLOGICAL CONDITIONS
Classical monocytes (M1) are a population of cells with diverse differentiation potential. They make up about 80%-95% of circulating monocytes. A distinctive feature of non-classical monocytes is that they have a gene expression program that allows them to migrate into tissues under normal conditions[14]. Normally, classical monocytes enter the bloodstream from the bone marrow and circulate for about a day, after which they replace some of the tissue macrophages in the intestines, heart, pancreas, and dermis[15] or form into non-classical monocytes. Under normal physiological conditions, classical monocytes make up a large proportion of the pool of tissue macrophages and are found in almost all tissues[16,17]. Adult monocytes, in some cases, can repeat the structure of resident macrophages while maintaining their identity[18] and may respond differently during inflammation[19]. There is a subpopulation of classic CD14bright/CD56+ monocytes that is less well characterized[15]. CD56+ monocytes proliferate in autoimmune diseases such as rheumatoid arthritis and Crohn’s disease. Being effective antigen-presenting cells, they can produce more reactive oxygen species and pro-inflammatory cytokines[19].
FORMATION OF NON-CLASSICAL MONOCYTES IN NORMAL AND PATHOLOGICAL CONDITIONS
Non-classical monocytes make up about 2%-11% of circulating monocytes. They have an excellent transcriptomic and metabolic profile (features of the respiratory chain enzyme complex) compared to classical monocytes, which use carbohydrate metabolism as an energy source[20]. Non-classical monocytes may have pro-inflammatory properties and secrete inflammatory cytokines in response to an infection[21]. They are involved in antigen presentation and T cell stimulation[22]. There is an increased content of non-classical monocytes in arthritis. Puchner et al. (2018), in an experiment on hTNFtg mice predisposed to arthritis, showed that the number of non-classical monocytes has increased at the preclinical stage[23]. Gazzito Del Padre et al. (2021) found that in patients with chronic inflammation of the blood vessels (Behcet’s disease), the number of circulating classic monocytes was lower and the number of intermediate monocytes was higher compared to apparently healthy donors[24].
INTERMEDIATE PHENOTYPE IN NORMAL AND PATHOLOGICAL CONDITIONS
Intermediate monocytes (M0) make up about 2%-8% of circulating monocytes[25]. Studies on human and other monocyte subpopulations have shown conflicting results; in one study, a closer association of intermediate monocytes with classical monocytes[26]: and in another with non-classical monocytes[27]. Their functions consist in the production of reactive oxygen species (ROS); they also present antigens, participate in proliferation and stimulate T cells. In addition, they are necessary for inflammatory reactions and angiogenesis[28]. It is unknown whether intermediate monocytes exist as a separate subgroup or are simply transitional stages between classical and non-classical monocytes[29]. Intermediate monocytes are common in bacterial sepsis, Crohn’s disease, cardiovascular disease[30], and rheumatoid arthritis[31]. Cormican et al. observed that intermediate blood monocytes can be sequentially classified into subpopulations with high and medium expression levels of the MHC II HLA-DR protein. They also showed that the number and proportions of subpopulations are regulated differently in different pathological conditions[32].
THE ROLE OF MITOCHONDRIA IN VARIOUS SUBPOPULATIONS OF MONOCYTES
Recent studies have shown that mitochondria not only produce adenosine triphosphate (ATP) but also participate in the regulation of calcium homeostasis, the formation of reactive oxygen species (ROS), and redox reactions and support the competence of immune cells. Furthermore, the focus will be on the role of mitochondria in monocytes in pathological conditions. Stimulation of monocytes by microbial ligands, such as lipopolysaccharide, peptidoglycan, or β-glucan, reprograms their metabolism to support the increased physiological demands that are necessary to generate an anti-inflammatory response[33]. The reprogrammed phenotype induces increased glycolysis and flux of the mitochondrial tricarboxylic acid cycle and oxidative phosphorylation[34].
For example, a group of scientists proved that LPS stimulation in monocytes causes changes in the morphology of mitochondria. They observed that the mitochondrial length was much shorter after 2 h of LPS treatment, indicating LPS-induced mitochondrial fragmentation in monocytes[35]. A change in the shape of mitochondria leads to an increase in membrane potential, the accumulation of succinate, and a shift in glycolysis. As a result, the production of ROS in mitochondria is increased, and monocytes produce pro-inflammatory cytokines.
In another study on monocytes from donors of atherosclerotic patients, researchers showed that the polarization of mitochondria, and, consequently, their functional state, can affect the pro-inflammatory activity of immune cells. They examined MMP (mitochondrial membrane potential) in various human monocyte subpopulations using MitoTracker on a flow cytometer. The proportion of monocytes with low levels of mitochondrial dye may be associated with the presence of atherosclerotic lesions in donors and a reduced ability of monocytes to secrete TNF in response to LPS[36]. The authors showed that non-classical monocytes demonstrate a reduced content of MMPs, increased production of reactive oxygen species (ROS), and shortened telomeres compared to classical monocytes. Another group studied dendritic cells of monocyte origin with impaired immunostimulatory ability. In such cells, the MMP content was reduced and signs of a pre-apoptotic state were found[37]. The results obtained allow us to conclude that the body level of mitochondrial depolarization can be associated with various pathologies that are characterized by inflammatory and metabolic disorders due to changes in the structure and functions of mitochondria. It is also interesting to note that the balance of energy metabolism in monocytes changes with age from oxidative phosphorylation to aerobic glycolysis. A decrease in mitochondrial potential leads to a decrease in the reserve respiratory capacity. Thus, the metabolic fitness of elderly monocytes appears to be impaired due to reduced mitochondrial respiratory reserve and a limited ability to utilize additional glucose[38]. All these data indicate the key role of mitochondria in the functioning of monocytes and the formation of a response to the impact of foreign agents.
MITOCHONDRIAL DYNAMICS IN MONOCYTES ARE NORMAL
At the same time, differences were found in glucose metabolism in monocytes/macrophages of various phenotypes depending on their activation. This is essential for macrophages to perform their functions. For example, macrophage activation by LPS induces a classically activated (M1) pro-inflammatory macrophage phenotype in which there is an increase in glycolysis and disruption of the Krebs cycle to support cellular metabolism and produce cytokines. Stimulation of macrophages by IL-4 leads to the activation of an alternative (M2) phenotype of anti-inflammatory macrophages. In this case, cells use fatty acid oxidation (FAO) and oxidative phosphorylation for cellular metabolism and the formation of ATP[39]. FAO is the main mechanism in cell metabolism for the production of ATP.
Lipid metabolism is very important for macrophage function, and it is possible that its deficiency may affect macrophage activation. Geric et al. found that the classical activation of macrophages disrupts mitochondrial production, and alternative activation weakly induces it[40]. Moreover, Zhu et al. reported that FAO inhibition can reduce the differentiation of monocytes into macrophages in vitro and in vivo[41].
In response to stress, mitochondria in monocytes try to maintain their normal composition and structure through the action of antioxidants, DNA repair, protein folding, and degradation[42]. Mitochondria often change their shape under different conditions. In particular, mitochondrial dynamics regulate the interconnection of the mitochondrial network, which depends on the metabolic needs of the cell[43]. The processes that control the dynamics of mitochondria have been well studied. It is known that the state of the dynamic network depends on the balance between fusion and fission, which are regulated by specific proteins[44]. The cell’s developmental status, metabolism, and microenvironment can upset the balance between these processes. An important feature of mitochondrial dynamics is that it regulates the functions of macrophages[45]. For example, M1 macrophages are known to contain fragmented mitochondria due to an increase in mitochondrial division caused by DRP1 activation, which leads to increased glycolysis[46]. This process is also associated with mitophagy, an event that follows the division of mitochondria in macrophages[47]. Furthermore, M2 macrophages have elongated mitochondria with a high level of FAO and oxidative phosphorylation due to mitochondrial fusion[48]. Thus, M1 macrophages show more signs of active mitophagy than M2 macrophages.
MITOCHONDRIAL FUSION PROTEINS
The process of mitochondrial fusion is regulated by the Misato protein, which is encoded by the MSTO1 gene that is expressed in all cells. It is localized both to the outer mitochondrial membrane and in the cytoplasm[49]. Mitochondrial fusion includes the fusion of MOM and MIM[50]. Both of these processes are regulated by members of the GTPase family. MOM fusion is regulated by mitofusin 1 (MFN1) and mitofusin 2 (MFN2), which undergo homotypic and heterotypic interactions in adjacent mitochondria[51]. Misato (MSTO1) interacts with MOM and promotes mitochondrial fusion[52]. MOM fusion is, in turn, regulated by MFN1 and MFN2[53]. MIM fusion is mediated by the optic atrophy protein 1 (OPA1), which is expressed in different isoforms through alternative splicing and protease cleavage. The fusion of MOM and MIM occurs almost simultaneously, which allows mitochondria to exchange their contents[54], and this process also protects dysfunctional mitochondria from mitophagy. GTPases, which are located in the outer membrane of mitochondria, control mitochondrial fusion by forming homodimeric or heterodimeric, antiparallel, helical bonds between neighboring mitochondria and C-terminal domains[54]. The deficiency of MFN1 and MFN2 leads to reduced mitochondrial fusion[55]. In addition, MFN2 controls cellular apoptosis and mitochondrial autophagy[56]. OPA1, a mitochondrial fusion protein, may also play a role as mitophagic factors. BNIP3 induces mitochondrial fragmentation through interaction with OPA1.
MITOCHONDRIAL DIVISION PROTEINS
Mitochondrial division is regulated by a dynamin-related protein (DRP1), GTPase; since this protein is localized in the cytosol, specific adapter proteins are required for its anchorage in the MOM[57]. These adapter proteins include fission protein (FIS1), mitochondrial fission factor (MFF), mitochondrial elongation factor (MIEF1/MID51), and mitochondrial elongation factor (MIEF2/MID49)[58]. DRP1 forms a ring-like structure around mitochondria by self-assembly[59]. DRP1 acetylation contributes to cardiomyocyte death and dysfunction, which are associated with metabolic stress[60]. Drp1-mediated mitochondrial division is also required for phagocytes to remove apoptotic cells, thus reducing necrosis and inflammation[61]. The process of division of mitochondria is completed by dividing them into small and fragmented ones. This plays a key role in cell division, as the resulting daughter cells have the same number of mitochondria as the mother cells. Moreover, the division promotes mitochondrial transport and the separation of damaged mitochondria[62]. Thus, mitochondrial fusion contributes to the expansion of mitochondrial networks, which gives an advantage to cells at high energy costs, and a violation of mitochondrial fusion leads to mitochondrial dysfunction[14].
REGULATORS OF MITOCHONDRIAL DYNAMICS
As mentioned above, mitochondrial fusion in mammals is controlled by MFN1 and MFN2, proteins that are localized on the outer mitochondrial membrane[63]. In addition, MFN2 is involved in the modulation of energy metabolism, the interaction of the endoplasmic reticulum and mitochondria, and the regulation of mitophagy[64]. Another component that regulates mitochondrial fusion is OPA1, a transmembrane protein associated with the inner mitochondrial membrane[65]. OPA1 undergoes proteolytic processing, resulting in the short (s) and long (l) isoforms, both required for mitochondrial fusion[66]. Mitochondrial division in mammalian cells is controlled by GTPases DRP1, FIS1, mitochondrial fission factor (MFF), and 49 and 51 kDa mitochondrial dynamic proteins[67]. DRP1 acts as a mechano-enzyme that is needed to physically constrict mitochondria during early division. It is noteworthy that DRP1 lacks a mitochondrial destination sequence; therefore, for the formation of the fission complex, FIS1 must be located on the outer mitochondrial membrane. A slight decrease in the level of MFF leads to elongation of mitochondria and reduces the translocation of DRP1 into mitochondria[68].
MITOCHONDRIAL TRANSPORT
Mitochondrial transport is carried out through microtubules and actin filaments in the cytoplasm of cells[69]. The movement of mitochondria requires the motor proteins myosin, kinesin, and dynein, which bind MOM proteins to cytoskeletal structures, forming a complex[70]. The division of mitochondria facilitates their movement since, in the cytoplasm, small and fragmented mitochondria are transported better than large and fused ones[71]. Mitochondrial transport promotes the subcellular localization of mitochondria, which is important to ensure the distribution of mitochondria during cell division and to maintain the energy needs of all cell types.
MITOCHONDRIAL DYNAMICS IN METABOLIC AND SOME INFLAMMATORY DISORDERS
We herein emphasize that mitochondrial fusion and fission are closely related to mitochondrial function. ROS are a byproduct of mitochondrial respiration since, when the level of mitochondrial ROS (mROS) is disturbed, processes occur that are involved in the development of inflammatory and metabolic disorders, such as atherosclerosis[72], diabetes, and obesity[73]. Interestingly, disturbances in mitochondrial metabolism, which is regulated by mitochondrial proteins Opa1 and Drp1 in various tissues, lead to the development of metabolic diseases[74].
High-fat and/or -sucrose diets can lead to insulin resistance (IR) and are often associated with mitochondrial dysfunction. Excess fatty acids or sucrose (glucose plus fructose) significantly overload the cell[75]. In the context of obesity, adipocytes play an important role in balancing metabolic homeostasis in response to excess energy. On a high-fat diet, mitochondria prefer a fragmented architecture associated with reduced adenosine 5’-triphosphate (ATP) production efficiency and increased release of reactive oxygen species (ROS)[76]. In obesity, adipocytes undergo hypertrophy, promoting the release of free fatty acids (FFA) during lipolysis and the development of hypoxia in adipose tissue. Necrotic adipocytes are phagocytosed by macrophages that produce chemokines and pro-inflammatory mediators[77]. Immune cells and macrophages are recruited into adipose tissue by inflammatory mediators [Figure 1].
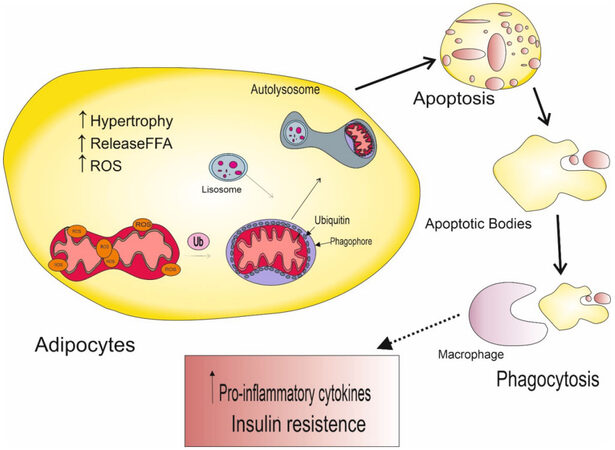
Figure 1. Pathological effects of a high-fat diet on mitochondrial function in adipocytes.
Oxidative stress is characterized by an imbalance between ROS production and the antioxidant capacity of defense systems. Mitochondrial function is required for ATP synthesis to support adipocyte energy requirements for processes such as lipid metabolism (tricarboxylic acid cycle and β-oxidation) and adipocyte differentiation and maturation[78]. Prolonged exposure to oxidative stress leads to oxidative DNA damage, causing mitochondrial dysfunction and disruption of fusion and fission processes, which ultimately lead to a vicious cycle causing lipid accumulation and insulin resistance[79]. Violation of β-oxidation contributes to the accumulation of lipids in the cell, resulting in an increase in the number of active lipid intermediates such as diacylglycerols (DAG) and ceramides (CER), which can inhibit the action of insulin. Decreased substrate oxidation and oxidative phosphorylation lead to decreased electron flow through the electron transport chain (ETC), which causes electron leakage and superoxide production, followed by oxidative stress and mitochondrial damage[80]. Mitophagy removes damaged mitochondria while maintaining normal mitochondrial homeostasis. Homeostasis shifts when mitophagy is disturbed, as non-functioning mitochondria cannot be removed. Because of this, oxidative stress increases and normal mitochondria are damaged. There are numerous studies confirming the critical role of mitochondrial dysfunction in the formation of various pathologies[81].
For example, Drp1 mutations lead to the development of vascular diseases through mechanisms such as myocardial ischemia-reperfusion (I/R), heart failure, and endothelial dysfunction in atherosclerosis[82,83]. Deletion of mitofusin 2 (MFN2), which regulates MOM fusion in conjunction with mitofusin 1 (MFN1) in the liver and skeletal muscle of mice, leads to fragmentation of mitochondrial networks, resulting in glucose intolerance and increased hepatic gluconeogenesis[84]. In brown adipose tissue (BAT), deletion of Mfn2 remodels mitochondrial dysfunction and increases insulin sensitivity and a predisposition to obesity[85]. Moreover, Mfn2 expression has been found to be significantly downregulated in the presence of atherosclerosis in ApoE-/- mice and is also involved in the pathogenesis of atherosclerosis[86,87]. All of these data support a critical regulatory role of Mfn2 in obesity and T2DM-associated glucose metabolism disorders[80] as well as in the development of inflammation in atherosclerosis[88].
In obesity, mitochondrial dynamics are disrupted. Mitochondrial dynamics depend on the availability of nutrients and energy consumption of cells[89]. It is known that, with nutrient deficiency and increased energy consumption, the mitochondrial network becomes elongated, while an abundant supply of nutrients and a decrease in energy requirements are associated with mitochondrial fragmentation[90]. Oma1 deficiency disrupts the mitochondrial fusion-fission balance and affects the reduction of oxidative phosphorylation, thereby enhancing fatty acid oxidation and reducing energy expenditure. The combination of these factors causes obesity in mice[91]. Tezze et al. showed that with age in people who lead a sedentary lifestyle, there is a decrease in Opa1, which is associated with loss of muscle mass[92]. Experiments on a mouse model of obesity caused by a high-fat diet have shown that the expression of MFN1 and MFN2 in skeletal muscles significantly decreases, and the expression of mitochondrial division mediators DRP1 and FIS1 increases, which leads to an increase in mitochondrial fission[93].
Mitochondrial dysfunction and its influence on the regulation of monocyte functioning
A common manifestation of inflammation is a change in mitochondrial structure and bioenergetics, which regulate MROs, enhance mitochondrial dysfunction, and promote inflammation and inflammatory cell death[94]. Disruption of the formation of mitochondrial ROS can cause cell damage and inflammatory death of macrophages, which is induced by oxidative stress[95]. Mitochondrial biogenesis and energy metabolism are necessary to regulate the inflammatory state of monocytes[96]. This condition is partially controlled by the peroxisome proliferator-activated gamma-receptor 1-alpha (PGC-1a) coactivator, which plays an important role in mitochondrial biogenesis by regulating genes involved in fatty acid oxidation and oxidative phosphorylation [Figure 2]. PGC-1a is expressed in endothelial cells, causing an increase in the expression of mitochondrial antioxidant enzymes[94].
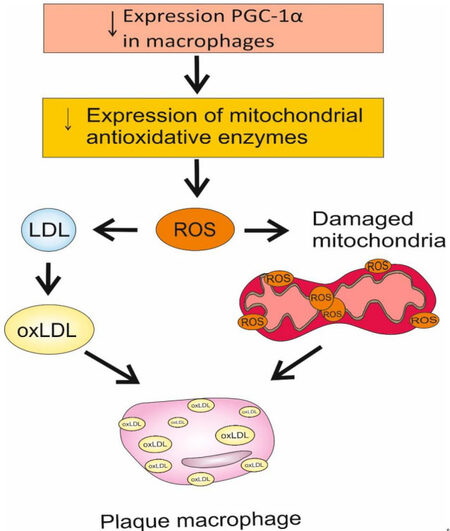
Figure 2. Pathogenesis of mitochondrial dysfunction and formation of foam cells in response to changes in PGC-1a expression.
PGC-1α is also found in macrophages of atherosclerotic plaque. It has been proven that its overexpression, when treated with conjugated linoleic acid (CLA), prevents the development of foam cells, preventing the capture of oxidized lipids by macrophages. Accordingly, a decrease in the expression of PGC-1a can lead to mitochondrial dysfunction and the formation of foam cells due to an increase in ROS production and a decrease in the action of antioxidants[96]. In addition, the function of mitochondria can be negatively affected by oxidized low-density lipoprotein (oxLDL) and ROS [Figure 2]. Damaged mitochondria release components, for example, mtROS, cardiolipin, and mtDNA, the action of which leads to inflammation due to increased production of interleukins 1ß and 18[95]. With impaired mitochondrial functions in monocytes/macrophages, chronic inflammatory diseases may develop, for example, pulmonary fibrosis, atherosclerosis[97], and rheumatoid arthritis[98]. For the regulation of inflammation, it is very important that mitochondrial processes are not disrupted in monocytes/macrophages[99].
There is now ample evidence pointing to mitochondrial dysfunction as a key mediator of cell death, such as neuronal death in Parkinson’s disease (PD)[100]. Smith et al. investigated mitochondrial pathology in peripheral monocytes of the control group and PD patients, as well as in lymphocytes and the total number of peripheral blood mononuclear cells (PBMC), by measuring the content of mitochondria (MitoTracker) and the production of mROS (MitoSox). They found that monocytes from PD patients had a significantly lower mitochondrial content and a higher level of mitochondrial ROS. In addition, during the analysis of the level of mROS by mitochondrial content (MitoSox/MitoTracker), higher levels of mitochondrial ROS were found in monocytes of patients with PD[101].
Williams et al. drew attention to the fact that patients with urolithiasis have reduced mitochondrial function in circulating monocytes compared to healthy people[102]. Scientists have suggested that this is due to the accumulation of oxalate in the renal epithelium. Perhaps macrophages are involved in the removal of salt crystals. Insufficient clearance may result in increased inflammation, oxidative stress, and tissue damage[103].
Shen et al. showed that, compared with wild-type mice, deficiency of receptor-associated tumor necrosis factor 3 (TRAF3), a member of the TRAF family with E3 ligase activity that functions as an important mediator of innate immunity receptor signaling, resulted in a significant decrease in production IL-1β and neutrophil infiltration. These data suggest that TRAF3 regulates mitochondrial ROS production, inflammation activation, and cell death[98].
Geisberger et al., in ex vivo and in vivo experiments, showed that macrophage CD36 receptor promotes reprogramming of mitochondrial metabolism from oxidative phosphorylation to ROS production to trigger chronic inflammation in atherosclerosis. Scientists have hypothesized that changing the sodium microenvironment could lead to a loss of mitochondrial function with reduced ATP production in all M1 and M2 macrophages, which could accelerate the development of cardiometabolic diseases[104].
Monocytes in obesity and inflammation: common features and differences
Recruited monocytes are involved in widespread tissue damage in both chronic and acute inflammation. During inflammatory reactions, secretion of inflammatory and anti-inflammatory cytokines occurs, which become key players in the development of a pathological condition[105]. Scientists have not yet been able to identify any single inflammatory cytokine as a specific biomarker. However, what is certain is that, in almost all inflammatory conditions, both in resolution and persistent inflammatory situations, there is a change in the profile of circulating cytokines[106]. This means that the acute and chronic phases largely overlap in terms of inflammatory cytokine levels, with differences occurring over time in the change in cytokine production. For example, in atherosclerosis, inflammation is observed with the accumulation of oxidized LDL in the outer and middle layer of the artery, and monocytes penetrate into the tissues, where they differentiate into macrophages[107]. Macrophages can change their inflammatory phenotype when exposed to the microenvironment[108]. Macrophages are divided into macrophages of the first type (M1) that have a pro-inflammatory activation pathway and macrophages of the second type (M2) that have an anti-inflammatory activation pathway[109]. M1 macrophages produce pro-inflammatory cytokines such as TNFa, IL-6, IL-8, and IL-1β[110]. M2 macrophages are responsible for the production of anti-inflammatory cytokines, for example, IL-1 (IL-1ra), IL-10, and CCL118[111]. For the production of inflammatory cytokines, the toll-like receptor (TLR) pathway can be activated to activate the NF-kB transcription factor system. In this case, pathogen-associated molecular patterns (PAMP) are used, such as lipopolysaccharide (LPS), flagellin obtained from bacteria, and a double-stranded viral RNA molecule [112]. Macrophages switch their phenotype to M1 during inflammation, depending on glycolysis for ATP synthesis, and to M2 during oxidative phosphorylation[113]. It is important to note that macrophages M1 and M2 are very plastic, moving from one form to the other. Glycolysis and oxidative phosphorylation occur in both phenotypes and activate the macrophage polarization pathway depending on the environment[114]. The phenotype of macrophages can switch depending on the oxygen level in the cells, and the oxygen level can also affect their metabolic processes[115]. When there is an increase in oxygen in tissues, peroxisome proliferation receptor-γ (PPAR-γ) begins to act in cells, while in mitochondria, there is a transition from aerobic glycolysis to oxidative phosphorylation[116]. As noted above, metabolites play an important role in the differentiation of monocytes and affect their functionality. For example, during prolonged fasting, short-chain fatty acid β-hydroxybutyrate is released from the liver and downregulates the inflammasome-induced NLRP3 IL receptor[117]. The number and composition of monocyte populations can also be influenced by eating behavior[118]. There are studies showing that with obesity, there is an increase in the number of intermediate and non-classical subsets of monocytes in circulating blood[93]. In obese patients, increased expression of TLR4 and TLR8 was observed in monocytes, and they also excessively secreted pro-inflammatory cytokines (IL-1ß and TNF) under the action of LPS[119,120]. In obesity, the function of monocytes changes due to various metabolites; similar processes are observed during inflammation in atherosclerosis. For example, the action of oxidized low-density lipoprotein (LDL) switches the phenotype of monocytes; they begin to increase the expression of pro-inflammatory chemokines[121,122]. Single nucleotide polymorphisms in the regions of the inflammatory adapter gene and IL-1 receptor antagonist have been shown to influence the response of human monocytes, suggesting that inflammation is involved in this process[123,124]. Restriction of calories and fatty foods has a beneficial effect on many chronic metabolic disorders such as T2DM2, non-alcoholic liver disease, and cardiovascular disease[125], and short-term fasting is sufficient to reduce the number of entire monocyte populations in healthy individuals[126]. To date, the mechanism of change in monocytes due to various dietary habits is not fully known, but modern technologies such as multicolor flow cytometry, mass cytometry, and single-cell RNA sequencing contribute to the understanding of the primary and secondary effects of diet on monocytes[127,128].
DISTURBANCES IN MITOCHONDRIAL DYNAMICS IN MONOCYTES AS A POTENTIAL DIAGNOSTIC TARGET IN DIAGNOSING AND PREDICTING THE COURSE OF METABOLIC AND INFLAMMATORY PATHOLOGICAL CONDITIONS
Due to the high prevalence of metabolic and inflammatory diseases, it is very important to develop a special approach to their diagnosis. This review describes the mechanisms of occurrence of these pathologies at the cellular level and proves that the disruption of mitochondrial dynamics plays a key role. There is a new diagnostic and treatment approach based on the use of photosensitivity, phototheranostics. This approach targets pathologically altered cells and tissues and only targets cellular organelles if the photosensitizing agent accumulates selectively. For example, such selectivity may result from damage to the mitochondrial genome. Scientists believe that such approaches can be applied in atherosclerotic diseases, when mitochondrial DNA mutations associated with atherosclerosis lead to mitochondrial dysfunction[129]. Phototheranostics is part of the medicine of the future; it is not only a new approach in diagnostics, but it can also be effective in subcellular therapy, including at the mitochondrial level.
Malik et al. proposed the hypothesis that the content of mtDNA in the tissues of the body may indicate alterations in mitochondrial functions. An increase in the amount of ROS as a result of external influences, for example, as a result of hyperglycemia or an increase in fat content under conditions of oxidative stress, can lead to an increase in mitochondrial biogenesis. The level of mtDNA plays an important role in the proper functioning of mitochondria; its changes can increase oxidative stress and cause inflammation. Changes in mtDNA levels are observed in peripheral blood mononuclear cells and can be used as a marker of pathological processes in tissues[130].
POTENTIAL OF MITOCHONDRIAL THERAPY IN OBESITY AND INFLAMMATION
Maintenance of mitochondrial function is an effective method in the treatment of damaged tissues and can alleviate the clinical manifestations of diseases[131]. Mitochondria may be an important target for the development of new drugs for cardiovascular diseases[132], and they are part of a promising strategy for the treatment of atherosclerosis and metabolic diseases, including obesity, by modulating mitochondria[133]. A series of experiments showed that cardiovascular diseases can be avoided by eliminating the consumption of fatty foods, correcting excess body weight, and controlling blood sugar levels. These recommendations can help avoid mitochondrial damage[134]. In addition, Schneeberger et al. reported that proopiomelanocortin (POMC) induces a specific deletion of Ms2, which may lead to resistance of the endoplasmic reticulum (ER) to leptin and reduce the risk of obesity[135].
Mitochondrial antioxidants are currently in the preclinical and clinical stages of testing[136]. Ingredients of natural origin with antioxidant action are also being developed now[137]. Studies on ilexgenin A have shown that it has the ability to inhibit the expression of Drp1 and excessive mitochondrial division; it also reduces the production of ROS and inflammatory factors, thereby reducing inflammation in atherosclerosis[138,139].
Kumar et al. suggested that deletion of the CD36 gene in macrophages could provide a potential strategy to combat chronic inflammation in atherogenic conditions by blocking mitochondrial fatty acid import with Etogooxira, which attenuates mitochondrial ROS as well as inflammation activation[140].
The role of fatty acid oxidation in monocyte activation and differentiation is discussed above. It is possible that the regulation of β-oxidation may be useful in the fight against inflammation in atherosclerosis[141]. Scientists have suggested that inhibition of CAO by trimetazidine in macrophages reduces the secretion of the pro-inflammatory cytokine IL-1β[142]. This approach could help slow the progression of atherosclerosis.
CONCLUSION
Mitochondria form a dynamic network that interacts with all cellular components and is involved in the organization of various physiological processes and cellular responses to damaging factors. This review shows that changes in mitochondrial function are the main contributor to the development of inflammatory and metabolic disorders. Proper mitochondrial dynamics are necessary to maintain normal mitochondrial functions and homeostasis in the cell. It is necessary to understand the causal relationships that underlie the mechanisms of inflammation and obesity for further progress in the diagnosis and treatment of these pathologies. In addition, the control over the mechanisms that regulate the dynamics of mitochondria will prevent damage to them. Thus, disturbances in mitochondrial dynamics can serve as a good diagnostic target for identifying the initial stages of chronic and metabolic diseases, the mechanisms of which are based on impaired mitochondrial functions. Moreover, special attention should be paid to the study and development of therapy for these pathologies based on mitochondrial preparations. Targeting mitochondria can be an effective way to combat diseases associated with inflammation and metabolic disorders.
DECLARATIONS
Authors’ contributionsText preparation: Tolstik TV
Consultation, English improvement: Markin AM
Writing the manuscript, editing English: Tolstik TV, Markin AM
Editing: Bogatyreva AI
Manuscript editing: Grechko AV, Oishi Y
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe work was supported by the Ministry of Science and Higher Education of the Russian Federation, Project 122030200531-3 “Atherosclerotic lesion of the thoracic aorta with aneurysm”.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Fahed G, Aoun L, Bou Zerdan M, et al. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int J Mol Sci 2022;23:786.
2. Kawai T, Autieri MV, Scalia R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol 2021;320:C375-91.
3. Chen Y, Yang M, Huang W, et al. Mitochondrial metabolic reprogramming by CD36 signaling drives macrophage inflammatory responses. Circ Res 2019;125:1087-102.
4. Wu H, Wang Y, Li W, et al. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 2019;15:1882-98.
5. Supale S, Li N, Brun T, Maechler P. Mitochondrial dysfunction in pancreatic β cells. Trends Endocrinol Metab 2012;23:477-87.
6. Anderson EJ, Lustig ME, Boyle KE, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 2009;119:573-81.
7. Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res 2008;102:401-14.
8. Yoo SM, Jung YK. A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells 2018;41:18-26.
9. Srinivasan S, Guha M, Kashina A, Avadhani NG. Mitochondrial dysfunction and mitochondrial dynamics-the cancer connection. Biochim Biophys Acta Bioenerg 2017;1858:602-14.
10. Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal 2011;14:1939-51.
11. Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J 2021;40:e104705.
13. Guilliams M, Mildner A, Yona S. Developmental and functional heterogeneity of monocytes. Immunity 2018;49:595-613.
14. Calderon B, Carrero JA, Ferris ST, et al. The pancreas anatomy conditions the origin and properties of resident macrophages. J Exp Med 2015;212:1497-512.
15. Mossadegh-Keller N, Gentek R, Gimenez G, Bigot S, Mailfert S, Sieweke MH. Developmental origin and maintenance of distinct testicular macrophage populations. J Exp Med 2017;214:2829-41.
16. Schyns J, Bai Q, Ruscitti C, et al. Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat Commun 2019;10:3964.
17. Sawai CM, Babovic S, Upadhaya S, et al. Hematopoietic stem cells are the major source of multilineage hematopoiesis in adult animals. Immunity 2016;45:597-609.
18. T’Jonck W, Guilliams M, Bonnardel J. Niche signals and transcription factors involved in tissue-resident macrophage development. Cell Immunol 2018;330:43-53.
19. Bennett FC, Bennett ML, Yaqoob F, et al. A combination of ontogeny and CNS environment establishes microglial identity. Neuron 2018;98:1170-1183.e8.
20. Cronk JC, Filiano AJ, Louveau A, et al. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J Exp Med 2018;215:1627-47.
21. Grip O, Bredberg A, Lindgren S, Henriksson G. Increased subpopulations of CD16(+) and CD56(+) blood monocytes in patients with active Crohn’s disease. Inflamm Bowel Dis 2007;13:566-72.
22. Schmidl C, Renner K, Peter K, et al. FANTOM consortium. Transcription and enhancer profiling in human monocyte subsets. Blood 2014;123:e90-9.
23. Kapellos TS, Bonaguro L, Gemünd I, et al. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front Immunol 2019;10:2035.
24. Chimen M, Yates CM, McGettrick HM, et al. Monocyte subsets coregulate inflammatory responses by integrated signaling through TNF and IL-6 at the endothelial cell interface. J Immunol 2017;198:2834-43.
25. Puchner A, Saferding V, Bonelli M, et al. Non-classical monocytes as mediators of tissue destruction in arthritis. Ann Rheum Dis 2018;77:1490-7.
26. Gazzito Del Padre TC, Belem JMFM, de Aguiar MF, et al. Distribution of monocytes subpopulations in the peripheral blood from patients with Behçet’s disease - impact of disease status and colchicine use. Clin Immunol 2021;231:108854.
27. Boyette LB, Macedo C, Hadi K, et al. Phenotype, function, and differentiation potential of human monocyte subsets. PLoS One 2017;12:e0176460.
28. Cros J, Cagnard N, Woollard K, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 2010;33:375-86.
29. Sebastian A, Sanju S, Jain P, Priya VV, Varma PK, Mony U. Non-classical monocytes and its potential in diagnosing sepsis post cardiac surgery. Int Immunopharmacol 2021;99:108037.
30. Sampath P, Moideen K, Ranganathan UD, Bethunaickan R. Monocyte subsets: phenotypes and function in tuberculosis infection. Front Immunol 2018;9:1726.
31. Ziegler-Heitbrock L, Hofer TP. Toward a refined definition of monocyte subsets. Front Immunol 2013;4:23.
32. Cormican S, Griffin MD. Human monocyte subset distinctions and function: insights from gene expression analysis. Front Immunol 2020;11:1070.
33. Patil NK, Bohannon JK, Hernandez A, Patil TK, Sherwood ER. Regulation of leukocyte function by citric acid cycle intermediates. J Leukoc Biol 2019;106:105-17.
34. McBride MA, Owen AM, Stothers CL, et al. The metabolic basis of immune dysfunction following sepsis and trauma. Front Immunol 2020;11:1043.
36. Nikiforov NG, Ryabova A, Kubekina MV, et al. Two subpopulations of human monocytes that differ by mitochondrial membrane potential. Biomedicines 2021;9:153.
37. Kisand K, Peterson P. Metabolic fitness is decreased in monocytes of old individuals. Aging (Albany NY) 2020;12:18791-2.
39. Stunault MI, Bories G, Guinamard RR, Ivanov S. Metabolism plays a key role during macrophage activation. Mediators Inflamm 2018;2018:2426138.
40. Geric I, Tyurina YY, Krysko O, et al. Lipid homeostasis and inflammatory activation are disturbed in classically activated macrophages with peroxisomal β-oxidation deficiency. Immunology 2018;153:342-56.
41. Zhu Y, Dun H, Ye L, et al. Targeting fatty acid β-oxidation impairs monocyte differentiation and prolongs heart allograft survival. JCI Insight 2022;7:e151596.
42. Faas MM, de Vos P. Mitochondrial function in immune cells in health and disease. Biochim Biophys Acta Mol Basis Dis 2020;1866:165845.
43. Parikh SM, Yang Y, He L, Tang C, Zhan M, Dong Z. Mitochondrial function and disturbances in the septic kidney. Semin Nephrol 2015;35:108-19.
44. Bereiter-Hahn J, Vöth M. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc Res Tech 1994;27:198-219.
45. Xie JH, Li YY, Jin J. The essential functions of mitochondrial dynamics in immune cells. Cell Mol Immunol 2020;17:712-21.
46. Gao Z, Li Y, Wang F, et al. Mitochondrial dynamics controls anti-tumour innate immunity by regulating CHIP-IRF1 axis stability. Nat Commun 2017;8:1805.
47. Esteban-Martínez L, Sierra-Filardi E, McGreal RS, et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J 2017;36:1688-706.
48. Rambold AS, Pearce EL. Mitochondrial dynamics at the interface of immune cell metabolism and function. Trends Immunol 2018;39:6-18.
49. Wang H, Yi J, Li X, Xiao Y, Dhakal K, Zhou J. ALS-associated mutation SOD1G93A leads to abnormal mitochondrial dynamics in osteocytes. Bone 2018;106:126-38.
50. Cosentino K, García-Sáez AJ. MIM through MOM: the awakening of Bax and Bak pores. EMBO J 2018;37:e100340.
51. Bulthuis EP, Adjobo-Hermans MJW, Willems PHGM, Koopman WJH. Mitochondrial morphofunction in mammalian cells. Antioxid Redox Signal 2019;30:2066-109.
52. Gal A, Balicza P, Weaver D, et al. MSTO1 is a cytoplasmic pro-mitochondrial fusion protein, whose mutation induces myopathy and ataxia in humans. EMBO Mol Med 2017;9:967-84.
54. Böckler S, Chelius X, Hock N, et al. Fusion, fission, and transport control asymmetric inheritance of mitochondria and protein aggregates. J Cell Biol 2017;216:2481-98.
55. Arribat Y, Broskey NT, Greggio C, et al. Distinct patterns of skeletal muscle mitochondria fusion, fission and mitophagy upon duration of exercise training. Acta Physiol (Oxf) 2019;225:e13179.
56. Ciarlo L, Vona R, Manganelli V, et al. Recruitment of mitofusin 2 into “lipid rafts” drives mitochondria fusion induced by Mdivi-1. Oncotarget 2018;9:18869-84.
57. Odendall F, Backes S, Tatsuta T, et al. The mitochondrial intermembrane space-facing proteins Mcp2 and Tgl2 are involved in yeast lipid metabolism. Mol Biol Cell 2019;30:2681-94.
58. El-Hattab AW, Suleiman J, Almannai M, Scaglia F. Mitochondrial dynamics: biological roles, molecular machinery, and related diseases. Mol Genet Metab 2018;125:315-21.
60. Hu Q, Zhang H, Gutiérrez Cortés N, et al. Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ Res 2020;126:456-70.
61. Wu NN, Zhang Y, Ren J. Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxid Med Cell Longev 2019;2019:9825061.
62. Burman JL, Pickles S, Wang C, et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol 2017;216:3231-47.
64. Chen Y, Liu Y, Dorn GW 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 2011;109:1327-31.
65. Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol 2009;187:959-66.
66. Ban T, Ishihara T, Kohno H, et al. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol 2017;19:856-63.
67. Ikeda Y, Shirakabe A, Maejima Y, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 2015;116:264-78.
68. Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 2008;19:2402-12.
69. Wang X, An P, Gu Z, Luo Y, Luo J. Mitochondrial metal ion transport in cell metabolism and disease. Int J Mol Sci 2021;22:7525.
70. Quintana A, Hoth M. Mitochondrial dynamics and their impact on T cell function. Cell Calcium 2012;52:57-63.
72. Myasoedova VA, Di Minno A, Songia P, et al. Sex-specific differences in age-related aortic valve calcium load: a systematic review and meta-analysis. Ageing Res Rev 2020;61:101077.
73. Hu C, Shu L, Huang X, et al. OPA1 and MICOS Regulate mitochondrial crista dynamics and formation. Cell Death Dis 2020;11:940.
74. Baechler BL, Bloemberg D, Quadrilatero J. Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation. Autophagy 2019;15:1606-19.
75. Skuratovskaia D, Komar A, Vulf M, Litvinova L. Mitochondrial destiny in type 2 diabetes: the effects of oxidative stress on the dynamics and biogenesis of mitochondria. PeerJ 2020;8:e9741.
76. Jackisch L, Murphy AM, Kumar S, Randeva H, Tripathi G, McTernan PG. Tunicamycin-induced endoplasmic reticulum stress mediates mitochondrial dysfunction in human adipocytes. J Clin Endocrinol Metab 2020;105:2905-18.
77. Elorza AA, Soffia JP. mtDNA heteroplasmy at the core of aging-associated heart failure. An integrative view of OXPHOS and mitochondrial life cycle in cardiac mitochondrial physiology. Front Cell Dev Biol 2021;9:625020.
78. Wang J, Lin X, Zhao N, et al. Effects of mitochondrial dynamics in the pathophysiology of obesity. Front Biosci (Landmark Ed) 2022;27:107.
79. Lefranc C, Friederich-Persson M, Palacios-Ramirez R, Nguyen Dinh Cat A. Mitochondrial oxidative stress in obesity: role of the mineralocorticoid receptor. J Endocrinol 2018;238:R143-59.
80. Xu Z, Fu T, Guo Q, Sun W, Gan Z. Mitochondrial quality orchestrates muscle-adipose dialog to alleviate dietary obesity. Pharmacol Res 2019;141:176-80.
81. Drake JC, Wilson RJ, Laker RC, et al. Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. Proc Natl Acad Sci USA 2021;118:e2025932118.
82. Morales PE, Arias-Durán C, Ávalos-Guajardo Y, et al. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol Aspects Med 2020;71:100822.
83. Sobenin IA, Sazonova MA, Postnov AY, Bobryshev YV, Orekhov AN. Mitochondrial mutations are associated with atherosclerotic lesions in the human aorta. Clin Dev Immunol 2012;2012:832464.
84. Bach D, Naon D, Pich S, et al. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes 2005;54:2685-93.
85. Mahdaviani K, Benador IY, Su S, et al. Mfn2 deletion in brown adipose tissue protects from insulin resistance and impairs thermogenesis. EMBO Rep 2017;18:1123-38.
86. Chiong M, Cartes-Saavedra B, Norambuena-Soto I, et al. Mitochondrial metabolism and the control of vascular smooth muscle cell proliferation. Front Cell Dev Biol 2014;2:72.
87. Soldatov VO, Malorodova TN, Balamutova TI, Ksenofontov AO, Dovgan AP, Urozhevskaya ZS. Endothelial dysfunction: comparative evaluation of ultrasound dopplerography, laser dopplerflowmetry and direct monitoring of arterial pressure for conducting pharmacological tests in rats. RRP 2018;4:73-80.
88. Kulkarni SS, Joffraud M, Boutant M, et al. Mfn1 deficiency in the liver protects against diet-induced insulin resistance and enhances the hypoglycemic effect of metformin. Diabetes 2016;65:3552-60.
89. Li D, Yang S, Xing Y, et al. Novel insights and current evidence for mechanisms of atherosclerosis: mitochondrial dynamics as a potential therapeutic target. Front Cell Dev Biol 2021;9:673839.
90. Kyriakoudi S, Drousiotou A, Petrou PP. When the balance tips: dysregulation of mitochondrial dynamics as a culprit in disease. Int J Mol Sci 2021;22:4617.
91. Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011;13:589-98.
92. Quirós PM, Ramsay AJ, Sala D, et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J 2012;31:2117-33.
93. Tezze C, Romanello V, Desbats MA, et al. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab 2017;25:1374-1389.e6.
94. Liu R, Jin P, Yu L, et al. Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS One 2014;9:e92810.
95. Bhat S, Shrestha D, Massey N, Karriker LA, Kanthasamy AG, Charavaryamath C. Organic dust exposure induces stress response and mitochondrial dysfunction in monocytic cells. Histochem Cell Biol 2021;155:699-718.
96. Shen Y, Liu WW, Zhang X, et al. TRAF3 promotes ROS production and pyroptosis by targeting ULK1 ubiquitination in macrophages. FASEB J 2020;34:7144-59.
97. Sobenin IA, Salonen JT, Khasanova ZB, et al. Carotid atherosclerosis-related mutations of mitochondrial DNA do not explain the phenotype of metabolic syndrome. Vessel Plus 2019;3:14.
98. Sobenin IA, Sazonova MA, Postnov AY, Salonen JT, Bobryshev YV, Orekhov AN. Association of mitochondrial genetic variation with carotid atherosclerosis. PLoS One 2013;8:e68070.
99. Zhang X, Li X, Jia H, An G, Ni J. The m6A methyltransferase METTL3 modifies PGC-1α mRNA promoting mitochondrial dysfunction and oxLDL-induced inflammation in monocytes. J Biol Chem 2021;297:101058.
100. López-Armada MJ, Riveiro-Naveira RR, Vaamonde-García C, Valcárcel-Ares MN. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013;13:106-18.
101. Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci 2015;40:200-10.
102. Smith AM, Depp C, Ryan BJ, et al. Mitochondrial dysfunction and increased glycolysis in prodromal and early Parkinson’s blood cells. Mov Disord 2018;33:1580-90.
103. Williams J, Holmes RP, Assimos DG, Mitchell T. Monocyte mitochondrial function in calcium oxalate stone formers. Urology 2016;93:224.e1-6.
104. Patel M, Yarlagadda V, Adedoyin O, et al. Oxalate induces mitochondrial dysfunction and disrupts redox homeostasis in a human monocyte derived cell line. Redox Biol 2018;15:207-15.
105. Geisberger S, Bartolomaeus H, Neubert P, et al. Salt transiently inhibits mitochondrial energetics in mononuclear phagocytes. Circulation 2021;144:144-58.
106. Bajpai G, Bredemeyer A, Li W, et al. Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res 2019;124:263-78.
107. Italiani P, Mosca E, Della Camera G, et al. Profiling the course of resolving vs. persistent inflammation in human monocytes: the role of IL-1 family molecules. Front Immunol 2020;11:1426.
108. Poznyak AV, Nikiforov NG, Markin AM, et al. Overview of OxLDL and its impact on cardiovascular health: focus on atherosclerosis. Front Pharmacol 2020;11:613780.
109. Orekhov AN, Sobenin IA, Gavrilin MA, et al. Macrophages in immunopathology of atherosclerosis: a target for diagnostics and therapy. Curr Pharm Des 2015;21:1172-9.
111. Gratchev A, Schledzewski K, Guillot P, Goerdt S. Alternatively activated antigen-presenting cells: molecular repertoire, immune regulation, and healing. Skin Pharmacol Appl Skin Physiol 2001;14:272-9.
112. Gratchev A, Kzhyshkowska J, Duperrier K, Utikal J, Velten FW, Goerdt S. The receptor for interleukin-17E is induced by Th2 cytokines in antigen-presenting cells. Scand J Immunol 2004;60:233-7.
113. Zhang L, Wang C. Inflammatory response of macrophages in infection. Hepatobiliary & Pancreatic Diseases International 2014;13:138-52.
114. Rodríguez-Prados JC, Través PG, Cuenca J, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 2010;185:605-14.
115. Davis MJ, Tsang TM, Qiu Y, et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. mBio 2013;4:e00264-13.
116. Ravi S, Mitchell T, Kramer P, Chacko B, Darley-Usmar VM. Mitochondria in monocytes and macrophages-implications for translational and basic research. Int J Biochem Cell Biol 2014;53:202-7.
117. Vats D, Mukundan L, Odegaard JI, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 2006;4:13-24.
118. Youm YH, Nguyen KY, Grant RW, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med 2015;21:263-9.
119. Postat J, Bousso P. Quorum sensing by monocyte-derived populations. Front Immunol 2019;10:2140.
120. Poitou C, Dalmas E, Renovato M, et al. CD14dimCD16+ and CD14+CD16+ monocytes in obesity and during weight loss: relationships with fat mass and subclinical atherosclerosis. Arterioscler Thromb Vasc Biol 2011;31:2322-30.
121. Devêvre EF, Renovato-Martins M, Clément K, Sautès-Fridman C, Cremer I, Poitou C. Profiling of the three circulating monocyte subpopulations in human obesity. J Immunol 2015;194:3917-23.
122. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol 2014;34:1731-8.
123. Sobenin IA, Salonen JT, Zhelankin AV, et al. Low density lipoprotein-containing circulating immune complexes: role in atherosclerosis and diagnostic value. Biomed Res Int 2014;2014:205697.
124. Christ A, Günther P, Lauterbach MAR, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell 2018;172:162-175.e14.
125. Summerhill VI, Grechko AV, Yet SF, Sobenin IA, Orekhov AN. The atherogenic role of circulating modified lipids in atherosclerosis. Int J Mol Sci 2019;20:3561.
126. Wei M, Brandhorst S, Shelehchi M, et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci Transl Med 2017;9:eaai8700.
127. Jordan S, Tung N, Casanova-Acebes M, et al. Dietary intake regulates the circulating inflammatory monocyte pool. Cell 2019;178:1102-1114.e17.
128. Chistiakov DA, Revin VV, Sobenin IA, Orekhov AN, Bobryshev YV. Vascular endothelium: functioning in norm, changes in atherosclerosis and current dietary approaches to improve endothelial function. Mini Rev Med Chem 2015;15:338-50.
129. Sobenin IA, Sazonova MA, Postnov AY, Bobryshev YV, Orekhov AN. Changes of mitochondria in atherosclerosis: possible determinant in the pathogenesis of the disease. Atherosclerosis 2013;227:283-8.
130. Malik AN, Czajka A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013;13:481-92.
131. Chistiakov DA, Sobenin IA, Orekhov AN. Strategies to deliver microRNAs as potential therapeutics in the treatment of cardiovascular pathology. Drug Deliv 2012;19:392-405.
132. Markin AM, Markina YV, Sukhorukov VN, Khaylov AM, Orekhov AN. The role of physical activity in the development of atherosclerotic lesions of the vascular wall. Clin exp morphology 2019;8:25-31.
133. Zielonka J, Joseph J, Sikora A, et al. Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev 2017;117:10043-120.
134. Stanzione R, Forte M, Cotugno M, et al. Uncoupling protein 2 as a pathogenic determinant and therapeutic target in cardiovascular and metabolic diseases. Curr Neuropharmacol 2022;20:662-74.
135. Schneeberger M, Dietrich MO, Sebastián D, et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 2013;155:172-87.
136. Kiyuna LA, Albuquerque RPE, Chen CH, Mochly-Rosen D, Ferreira JCB. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic Biol Med 2018;129:155-68.
137. Myasoedova VA, Kirichenko TV, Melnichenko AA, et al. Anti-atherosclerotic effects of a phytoestrogen-rich herbal preparation in postmenopausal women. Int J Mol Sci 2016;17:1318.
138. Zhu Y, Li M, Lu Y, Li J, Ke Y, Yang J. Ilexgenin A inhibits mitochondrial fission and promote Drp1 degradation by Nrf2-induced PSMB5 in endothelial cells. Drug Dev Res 2019;80:481-9.
139. Soldatov VO, Malorodova TN, Pokrovskaya TG, et al. International journal of research in pharmaceutical sciences ultrasonic dopplerography for the evaluation of endothelial function in the conduct of pharmacological vascular samples in an experiment production and hosted by. Int J Res Pharm Sci 2018;9:735-40.
140. Kumar P, Patel M, Oster RA, et al. Dietary oxalate loading impacts monocyte metabolism and inflammatory signaling in humans. Front Immunol 2021;12:617508.
141. Puchenkova OA, Nadezhdin SV, Soldatov VO, et al. Study of antiatherosclerotic and endothelioprotective activity of peptide agonists of EPOR/CD131 heteroreceptor. Farm farmakol (Pâtigorsk) 2020;8:100-11.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Tolstik TV, Bogatyreva AI, Grechko AV, Oishi Y, Markin AM. Features of mitochondrial dynamics in monocytes in inflammatory and metabolic disorders. Vessel Plus 2022;6:58. http://dx.doi.org/10.20517/2574-1209.2022.22
AMA Style
Tolstik TV, Bogatyreva AI, Grechko AV, Oishi Y, Markin AM. Features of mitochondrial dynamics in monocytes in inflammatory and metabolic disorders. Vessel Plus. 2022; 6: 58. http://dx.doi.org/10.20517/2574-1209.2022.22
Chicago/Turabian Style
Tolstik, Taisiya V., Anastasia I. Bogatyreva, Andrey V. Grechko, Yumiko Oishi, Alexander M. Markin. 2022. "Features of mitochondrial dynamics in monocytes in inflammatory and metabolic disorders" Vessel Plus. 6: 58. http://dx.doi.org/10.20517/2574-1209.2022.22
ACS Style
Tolstik, TV.; Bogatyreva AI.; Grechko AV.; Oishi Y.; Markin AM. Features of mitochondrial dynamics in monocytes in inflammatory and metabolic disorders. Vessel Plus. 2022, 6, 58. http://dx.doi.org/10.20517/2574-1209.2022.22
About This Article
Copyright

Data & Comments
Data


 Cite This Article 23 clicks
Cite This Article 23 clicks

 Like This Article 25
likes
Like This Article 25
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.