
Disclosing the cardiomyopathic substrate within the heart muscles in amyloidosis by cardiac magnetic resonance: diagnostic and prognostic implications
 0
0Abstract
The use of cardiac magnetic resonance (CMR) for accurate morphological assessment of cardiomyopathies is well described. CMR tissue characterization with late gadolinium enhancement and parametric mapping is highly promising in differentiating key etiologies of left ventricular hypertrophy, diagnosing cardiac involvement in systemic amyloidosis, detecting early disease, and tracking changes over time, as well as providing valuable prognostic information. This review focuses on the typical imaging findings in cardiac amyloidosis by CMR, highlighting its potentials with respect to traditional imaging techniques. Furthermore, the diagnostic and prognostic role of CMR parameters and the future perspectives related to the newest applications are addressed.
Keywords
INTRODUCTION
Amyloidosis is caused by aggregation and deposition of misfolded autologous proteins in the extracellular space of several organs. Two types commonly infiltrate the heart: immunoglobulin light chain (AL) and transthyretin (ATTR) amyloidosis. Cardiac involvement is the leading cause of morbidity and mortality[1], occurring in approximately 80% of AL, with a median survival of six months in untreated patients[2]. Cardiac dysfunction results from infiltration, but there is also evidence for a toxic effect of light chains[3]. ATTR amyloidosis is most commonly from wild-type protein, with age-related misfolding (ATTRwt) and less commonly from variant TTR (ATTRv), caused by a germline mutation in ATTR gene. ATTRwt often involves the heart, with a prevalence around 15% in the seventh decade in patients with heart failure and preserved EF or in association with severe aortic stenosis[4-6]. ATTRv presents at an earlier age, following a varied clinical course depending on the specific mutation inherited, with either cardiomyopathy or polyneuropathy. ATTR has a median survival of 3-4 years from presentation in patients with predominantly cardiomyopathy[7,8].
Endomyocardial biopsy is the reference standard for diagnosing CA, with histopathological evidence of diffuse infiltration of the subendocardial layer and sometimes a patchy or transmural pattern. Immunoelectron microscopy, immunohistochemistry, or mass spectrometry can identify TTR and AL fibril types, and DNA analysis is used to differentiate ATTRv from ATTRwt, thus identifying causative mutations.
Besides limited availability, invasiveness, and requirement of expertise, a biopsy sample may not be representative of the entire heart, particularly in the early stages, with focal deposits[9]. The expert consensus statement for the diagnosis of cardiac amyloidosis[10] recommends a positive biopsy for amyloid fibrils. In the absence of endomyocardial biopsy-proven disease, cardiac amyloidosis can be diagnosed using a combination of extracardiac biopsy, Tc-PYP/DPD scintigraphy, myocardial uptake of targeted positron emission tomography amyloid tracers, echocardiographic, and/or CMR findings.
Very importantly, Tc-PYP/DPD scintigraphy consistent with ATTR cardiac amyloidosis combined with suggestive Echo or CMR findings obviates the need for invasive biopsy, allowing a clinical diagnosis of ATTR cardiac amyloidosis in the absence of serum and/or urine light chains[11]. Otherwise, the presence of a clonal plasma cell process makes a histological diagnosis still mandatory.
CA is an increasing focus for the imaging community, driven by expensive new therapeutic options, increasing survival in AL, and wider recognition of ATTR amyloidosis in the elderly; hence, early diagnosis of CA and typing of fibrils are critical.
Available imaging modalities offer substantial advantages for non-invasive diagnosis and estimation of amyloid burden, and they can be easily repeated in the follow-up, with complementary information.
The combination of ECG low voltages with LV thickening is highly suggestive for CA[12]. ATTR often shows more severe, asymmetric hypertrophy and systolic dysfunction as compared to AL, presenting with moderate symmetric hypertrophy, sometimes with non-hypertrophic LV[13]. Small ventricles, enlarged atria, diastolic dysfunction, right ventricular hypertrophy, thickened valves and interatrial septum, and pericardial and pleural effusions are also common.
Besides a fundamental role for the initial screening, echocardiography may be non-specific, particularly in earlier stages. Over the years, more specific echocardiographic features have been recognized, such as reduction in LV longitudinal strain (LS). Global LS is significantly impaired in CA even with normal EF[13], and AL-CA showed worse strain values as compared with ATTR-CA[13]. A regional pattern that spares the apex, giving the characteristic “bull’s-eye” appearance on strain polar maps, is sensitive (93%) and specific (82%) to distinguish CA from other causes of LVH[14]. However, although echocardiography can provide an assessment of the likelihood of cardiac amyloid infiltration, it cannot be considered a definitive diagnostic test.
CMR offers accurate information on cardiac structure and function, with the key advantage to provide information about myocardial tissue substrate. This is promising for crucial earlier diagnosis, better understanding of disease processes, and potential ability to track disease in response to treatment.
VALUE OF CMR IN AMYLOIDOSIS
Diagnosis
CMR is progressively becoming a standard of care in the diagnosis of CA. Beyond accurate routine evaluation of volumes, mass, and function, CMR feature tracking (CMR-FT) allowed a precise evaluation of more subtle functional impairment in CA. CMR confirmed the echocardiographic finding of a global reduction of LS, with a “relative apical sparing” pattern, also providing mechanistic insights: the concomitant presence of a base-to-apex gradient of late gadolinium enhancement (LGE) burden suggested that the regional strain gradient may be related to the burden of amyloid deposition[15]. This is paralleled by a recent 99mTc-DPD SPECT study in ATTR, showing less DPD uptake in the LV apex, reinforcing the concept of apical sparing from the pathophysiological standpoint[16].
Newer CMR sequence, i.e., fast strain-encoded magnetic resonance (fast-SENC), is an evolution of tagging technique, which may perform better than CMR-FT, allowing for a single heartbeat and more comprehensive evaluation of global myocardial strain, with high reproducibility. Fast-SENC demonstrated incremental value for the identification of patients with subclinical LV dysfunction due to non-ischemic cardiomyopathies, including CA[17]. Furthermore, the combination of atypical LGE and impaired strain, with a base-to-apex gradient by fast-SENC, demonstrated high accuracy for the differential diagnosis between HCM and CA[18].
However, the most important advantage of CMR is its unique non-invasive tissue characterization, which offers fundamental information on tissue composition, with histologic validation[19,20]. This is especially useful in patients with other causes of LV hypertrophy, differentiating CA from hypertensive heart disease, hypertrophic cardiomyopathy, or other infiltrative cardiomyopathies, which may be challenging by echocardiography[21]. Furthermore, CA can coexist with paradoxical low-flow, low-gradient aortic stenosis[6].
Two techniques are routinely adopted: LGE and parametric mapping imaging.
LGE imaging
The administration of chelated, gadolinium-based contrast agents, carried within the blood pool into the extracellular space of the myocardial tissue, allows differentiating between normal and abnormal myocardium. Gadolinium accumulates in the gaps between cells when the interstitial compartment increases, mainly as a consequence of cardiomyocytes necrosis with “replacement” fibrosis or primary abnormal protein deposition, as in CA.
The increased amount of gadolinium can be demonstrated applying the inversion recovery principle to T1-weighted imaging, with an adequate inversion time to null normal myocardium.
The most relevant feature of CA is the appearance of LGE images, where the blood pool is dark. This is a consequence of infiltration: myocardial extracellular and plasmatic volume tend to equalize, nulling approximately at the same time.
LGE imaging can be challenging in advanced stages with extensive involvement and almost no normal myocardium. The operator can erroneously null the abnormal myocardium, missing global infiltration or creating a false subendocardial sparing appearance; the development of phase sensitive inversion recovery (PSIR) sequences has made LGE imaging more robust and operator independent, overcoming this limitation[22].
Global subendocardial enhancement and transmural LGE are typical features of CA, with a sensitivity of 86% and a specificity of 92%[23], and increasing LGE extent shows good correlation with the degree of myocardial infiltration. LGE presents an initial basal predilection, progressing to biventricular transmurality in advanced disease, whereas early stages of CA can present with focal or even absent LGE, making the diagnosis more challenging[24] [Figure 1]. Finally, LGE is highly prevalent and more extensive in ATTR than AL, but with similar progression and significant overlap between the subtypes[22,24,25].
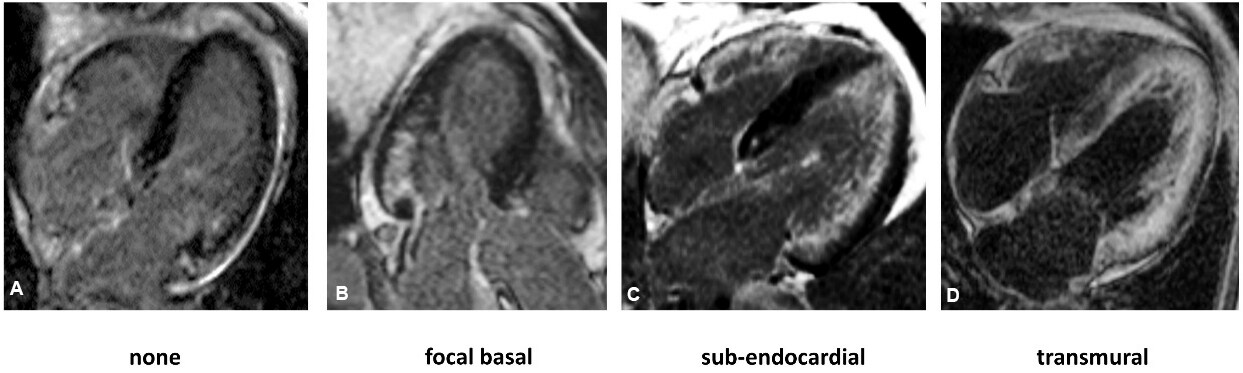
Figure 1. Late gadolinium enhancement imaging showing progression from early to advanced stages of cardiac involvement in amyloidosis. CMR images of four patients with cardiac AL amyloidosis: (A) early disease with no LGE; (B) early disease with focal basal LGE; (C) intermediate disease with subendocardial LGE; and (D) advanced disease with transmural LGE. LGE: Late gadolinium enhancement.
T1 and T2 mapping imaging
T1 mapping is a relatively new technique, where a direct quantitative signal from the myocardium is measured. Each pixel in the image is coded in color, reflecting the absolute T1 values in milliseconds. Pre-contrast T1 measures intrinsic signal from the myocardium due to its composition (native myocardial T1). Pathological processes changes T1, with reduced values in iron or fat infiltration, such as Fabry disease. Native T1 increases modestly in diffuse fibrosis, more in scar, and substantially in amyloid, being as high as 5 SD or higher than normal, often allowing a differential diagnosis vs. other hypertrophic phenotypes[21,26,27] [Figure 2].
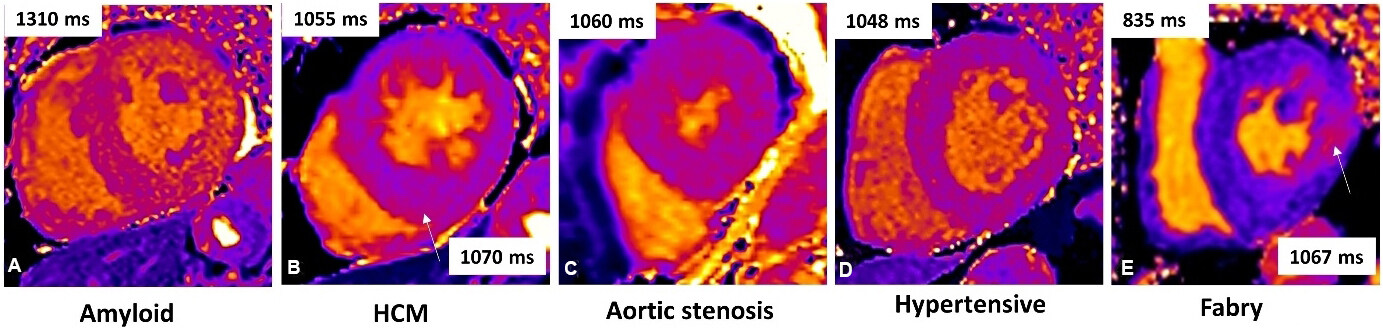
Figure 2. Cardiac magnetic resonance (CMR) native myocardial T1 by MOLLI T1 maps in different cardiac disease with LV hypertrophy: (A) amyloid with patchy native T1 elevation; (B) sarcomeric hypertrophic cardiomyopathy (HCM) with global mild T1 elevation and higher T1 in the infero-septum; (C) aortic stenosis with mild global T1 increase; (D) hypertensive heart disease with high-normal T1; and (E) fabry disease shows diffuse low T1 and pseudo-normal T1 in the inferolateral wall.
Native T1 presents high diagnostic accuracy for CA in both AL and ATTR with high pre-test probability, and it has been validated as a surrogate marker of infiltration, with good correlation with disease severity and markers of systolic and diastolic dysfunction[28,29]. Furthermore, native T1 demonstrated high accuracy in patients with suspected CA with various degrees of kidney dysfunction, showing very high positive and negative predictive values for extremely high and low T1 values, respectively[30].
In patients with eGFR < 30 mL/min/1.73 m2, it is generally suggested to avoid contrast administration, although currently used macrocyclic agents show a very good safety profile; in this setting, an individual assessment of the risk-to-benefit ratio should be considered, taking into account that CA could be detected by native T1 in a proportion of patients with advanced renal failure avoiding contrast, as well as reducing costs and time[30]. However, native T1 showed limited accuracy in patients with intermediate values. Furthermore, native T1 is a composite signal from both the extra- and intracellular space and cannot fully differentiate the underlying processes, particularly edema and amyloid[30].
After gadolinium administration, using the ratio of pre- to post-contrast T1 and hematocrit, the signal from the extracellular space can be isolated with the calculation of the extracellular volume (ECV), representing purely the extracellular expansion, which is the closest quantifier of amyloid burden within the heart.
Amyloidosis is the exemplar interstitial disease, and this is reflected by massive ECV elevation, up to 0.50-0.60 in definite CA. While ECV in healthy subjects is 0.22-0.28, it increases hugely in focal scar or edema, but, for diffuse fibrosis (aortic stenosis and hypertensive heart disease), the increases are 0.30-0.35.
A global ECV increase around 0.40 should raise the suspicion of CA. In fact, ECV showed a tight incremental correlation with probability of cardiac involvement based on echocardiographic and bio-humoral findings in AL[31], and it agreed well with ECV measured by histology, representing a direct marker of the amyloid burden[20].
Furthermore, ECV tracked a wide variety of markers of disease activity, such as cardiac function, blood biomarkers, and functional status[22,31,32]. Not surprisingly, ECV presented the best diagnostic accuracy for CA vs. other CMR parameters[33]; thus, unless gadolinium is contraindicated, ECV should be measured for a more definitive diagnosis, especially if there is mildly elevated native T1.
ECV measurements can also be applied to extracardiac organs, and, in AL amyloidosis, ECV tracked measures of amyloid burden assessed by Serum amyloid P scintigraphy[34]. With a multiparameter tissue characterization by CMR, some differences between AL and ATTR have been observed. This could reflect differing physiopathology, but it should not be used to differentiate cardiac amyloid subtype. Besides a more prevalent sub-endocardial LGE in AL than ATTR (more often transmural), AL patients present a greater elevation of native T1 and lower ECV, suggesting that the amyloid burden might be higher in ATTR; hence, higher native T1 with smaller amyloid burden in AL points to additional pathologic processes, and the most likely is edema related to light chain toxicity or rapid deposition[35-37]. Finally, preliminary observations of an increase of myocyte volume (1 - ECV × LV mass) in ATTR as compared to AL hypothesized a different myocyte response, with a possible protective hypertrophy in ATTR but myocyte loss through a toxic effect in AL.
T2 mapping sequences, able to quantify myocardial edema, showed higher mean T2 values in both AL and ATTR[37]. Increased myocardial T2 values correlated with the presence of edema on histology, in the absence of significant inflammation[37]. Patients with untreated AL amyloidosis showed the greatest increase in myocardial T2, but there was substantial overlap of T2 values between subtypes. Figure 3 shows a comprehensive CMR assessment of patients with cardiac amyloidosis.
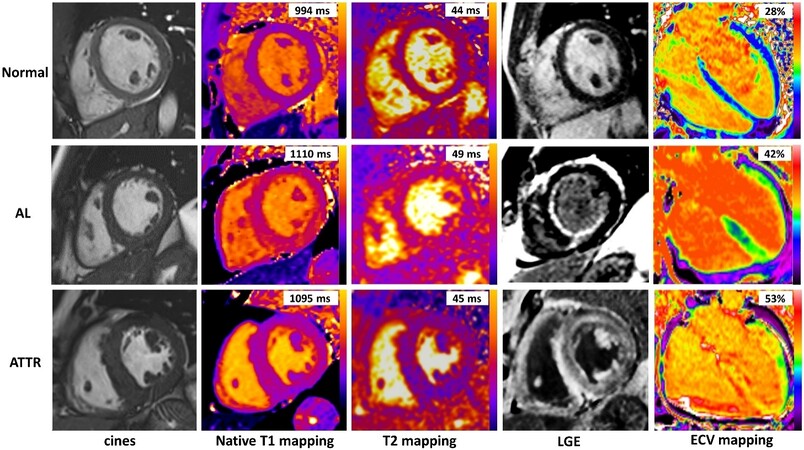
Figure 3. Multiparameter tissue characterization by cardiac magnetic resonance (CMR) in amyloid patients. CMR with cines, native T1 mapping, T2 mapping, late gadolinium enhancement (LGE), and extracellular volume (ECV) mapping in a normal subject (top row) and two patients with cardiac amyloidosis (middle and bottom rows) are shown. In the top row, the normal subject has no LGE and normal native T1, T2, and ECV. In the middle row, the patient with cardiac AL amyloidosis has high native T1, mildly increased T2, diffuse sub-endocardial LGE, and increased ECV values. In the bottom row, the patient with cardiac ATTR amyloidosis has high native T1, normal T2, transmural LGE, and very high ECV values.
Due to its unique tissue characterization, CMR has a highly important role in the diagnostic evaluation of suspected cardiac amyloidosis, in fact, the addition of LGE and mapping imaging can significantly contribute to diagnosis of AL CA, especially when echocardiography is not conclusive. In ATTR patients, bone scintigraphy Perugini Grade 2 or 3 with a suggestive echocardiogram is often diagnostic, but in uncertain cases CMR examination is recommended and could be useful to detect early disease[22,31,32,38,39].
STAGING OF CARDIAC INVOLVEMENT
Staging of cardiac involvement
Staging of CA is currently based on echocardiography and blood biomarkers, with limited accuracy when confounders (hypertension, renal failure, and atrial fibrillation) are present[40-42]. Furthermore, none of these parameters quantify the amyloid burden.
CMR explores the continuum of amyloid infiltration, showing correlation with clinical, bio-humoral, and morpho-functional parameters, possibly representing a useful tool for early detection and stratification of cardiac involvement.
First, LGE can be present in a proportion of patients in which routine assessment is not suggestive for CA[38], contributing to earlier detection of CA, but it can also be focal or even absent in early stages. With disease worsening, LGE transmural extent increases, and this is paralleled by higher T1 and ECV values[31,32,43].
Moreover, a limitation of LGE imaging is that it is not a quantitative marker in amyloidosis, owing to often diffuse infiltration, thus limiting its capability to precisely track changes of myocardial substrate. Quantitative T1 mapping overcomes these limitations, being an easy quantitative tool and more sensitive to early disease than LGE.
Native T1 is very high in advanced disease, it is less in early stages, and, in this setting, ECV estimation appears to be the most accurate parameter. ECV identified early CA in AL with values of ~0.40 and no other evidence of cardiac involvement (no LGE, wall thickening, or biomarker elevation). This was also shown in ATTR, where ECV was increased in a proportion of patients with possible ATTR (Perugini Grade 1) and no LGE[31,32,43].
The accuracy and precision of ECV measurements in the evaluation of substrate changes in the individual patient suggest the potential role of this marker for staging purposes.
Some limitations of parametric mapping need to be highlighted. Different CMR systems, field strength, and T1 mapping sequences have different normal ranges. Current recommendations are for normal reference ranges to be defined locally and to perform phantoms quality control regularly at each center. Although ECV demonstrated lower dependence on field strength, sequence choice, and imaging parameters, this is an obstacle for the implementation of mapping in clinical practice. Further work is required to obviate the need for local reference ranges, enabling the establishment of T1 and ECV reference standard to facilitate multicenter studies[44].
Prognosis
Currently, risk stratification of patients with AL and ATTR amyloidosis relies on biomarkers (NT-pro-BNP, troponin, free light chains, and eGFR) and clinical assessment, able to give prognostic information at presentation. Promising results derive from the study of the prognostic impact of changes of biomarkers, to assess disease progression and response to therapies[40,45].
CMR could add important prognostic information to the current clinical management.
Despite the excellent discriminative capacity of LGE, conflicting results were initially reported on prognostication. With the transition to more robust PSIR sequences, LGE imaging demonstrated the ability to predict mortality[22,46], as well as provide evidence of its progression from normal to subendocardial and transmural extent. Transmural LGE was an independent marker of all-cause mortality: two-year survival with no LGE was 92% in AL and ATTR, dropping to 81% with subendocardial LGE and 45% in AL and 65% in ATTR with transmural LGE[22,47].
As concerns the mapping technique, native T1 > 1044 ms and ECV > 0.45 predicted mortality at two years in AL amyloidosis, but the latter emerged as the stronger and independent predictor of mortality[31]. In a study comparing T1 mapping with LGE, ECV > 0.44 and global transmural LGE were independently prognostic; furthermore, in similar LGE patterns, ECV remained prognostic, while native T1 was not found to predict mortality[33,48].
Similarly, in ATTR, ECV and native T1 predicted mortality at 2.5 years. However, ECV showed stronger correlations with systolic and diastolic function and ECV ≥ 0.59 was associated with prognosis after adjustment for known independent predictors, including DPD grade and LGE, whereas myocardial T1 was not[25]. Finally, ECV demonstrated higher hazard ratio for adverse events compared with LGE and native T1 in a metanalysis, suggesting this metric as a robust prognostic marker[33].
The higher prognostic ability of ECV compared with LGE likely springs from its better discriminatory power, being able to measure the continuum of infiltration rather than a binary categorization. Furthermore, as compared with native T1, ECV likely reflects a purer quantifier of amyloid burden, not being influenced by intracellular content.
T2 mapping techniques were also demonstrated to be helpful in prognostication; myocardial T2 > 55 ms emerged as an independent prognostic factor after adjustment for ECV and NT-pro-BNP in AL patients, but not in ATTR patients[37]. This is possibly because of the additional toxic effect of light chains, which could contribute to cell death, inducing myocardial edema[3]. Prospective follow-up studies assessing changes during therapy are needed to verify the hypothesis. Table 1 shows the current role of CMR parameters for diagnosis, prognosis, and pathophysiological assessment in cardiac amyloidosis.
The current role of CMR parameters for diagnosis, prognosis, and pathophysiological assessment in cardiac amyloidosis
| Diagnosis | Prognosis | Physiopathology | |
| Morphology and function | + | + | + |
| Native T1 | ++ | ++ | ++ |
| T2 | - | + | ++ |
| ECV | +++ (early) | +++ | ++ |
| LGE | +++ | +++ | + |
Future perspectives
The evolving field of CMR techniques pointed out some new insights into pathophysiology of CA. Myocardial perfusion defects have been observed in CA, with different factors potentially contributing to hypoperfusion and cell damage (physical presence of amyloid deposits, infiltration of vessels and perivascular regions, and myocyte hypertrophy)[49,50]. Preliminary data using adenosine stress with myocardial blood flow (MBF) assessed by CMR perfusion mapping show severe reduction in stress MBF in CA, similar to patients with three-vessel coronary artery disease. The reduction correlates with the degree of amyloid infiltration and markers of adverse prognosis, being significant also in early disease[50]. Myocardial perfusion reserve impairment was also shown by stress N-13 ammonia positron emission tomography in both AL and ATTR, with coronary microvascular function inversely correlating to increased LV mass, diastolic filling pressures, and subclinical systolic dysfunction by longitudinal strain[49].Over the last decade, chemotherapy regimens for AL have improved, and several patients can now achieve large reductions in circulating light chains[51]. Multiple agents have been approved or are in late-phase trials for ATTR amyloidosis, as well[52]. NT-pro-BNP and echocardiography (more recently speckle tracking strain) are commonly used for assessing cardiac response to therapy, but they represent processes downstream of amyloid deposition. In contrast, ECV measurement by CMR has the potential to track structural changes (amyloid burden and cardiomyocyte response). Preliminary studies in AL showed that the decrease in ECV (and, to a lesser extent, LV mass and LGE) was higher in patients with complete or very good partial hematological response. Of note, a proportion of patients with good hematological response did not show ECV reduction[53].
Remarkable recent therapeutic developments in treating ATTR amyloidosis have been associated with improved outcome in patients with ATTR cardiomyopathy and polyneuropathy. Convincing evidence of cardiac response by echocardiography has not been demonstrated in patients treated with tafamidis[54], whereas, with patisiran, only a marginal improvement was observed[55]. Instead, data on serial CMR examinations with ECV suggest that regression of amyloid burden can occur in a proportion of patients treated with patisiran and diflunisal[56]. Although there was an average reduction of 6% in ECV compared to controls, individual responses varied substantially, highlighting the importance of an individual assessment of myocardial changes.
Tracking changes of the cardiac substrate has the potential to redefine cardiac response to treatment, allowing the discrimination of patients with lower risk of progression and patients with the need to intensify or change therapy. Furthermore, the ability to measure the amyloid burden could be of value as an endpoint in clinical trials for the development of new therapies.
CONCLUSION
CMR assessment provides a detailed morpho-functional analysis and a unique tissue characterization of the myocardial substrate in CA with prognostic value. Furthermore, CMR quantitative mapping is a promising tool for early diagnosis and monitoring of myocardial response to therapies.
DECLARATIONS
Authors’ contributionsConception, design and writing of the manuscript: Pica S
Revision the manuscript critically and approval the final manuscript submitted: Lombardi M
Availability of data and materialsThe data underlying this article will be shared on reasonable request to the corresponding author.
Financial support and sponsorshipThis study was in part supported by the Ministry of Health, Italy.
Conflicts of interestBoth authors report no potential conflicts of interest with Industries or other Institutions that could be construed as a conflict of interest related to the present work.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation 2009;120:1203-12.
2. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992;79:1817-22.
3. Brenner DA, Jain M, Pimentel DR, et al. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res 2004;94:1008-10.
4. González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015;36:2585-94.
5. Bennani Smires Y, Victor G, Ribes D, et al. Pilot study for left ventricular imaging phenotype of patients over 65 years old with heart failure and preserved ejection fraction: the high prevalence of amyloid cardiomyopathy. Int J Cardiovasc Imaging 2016;32:1403-13.
6. Castaño A, Narotsky DL, Hamid N, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J 2017;38:2879-87.
8. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid 2016;23:209-13.
9. Pellikka PA, Holmes DR Jr. , Edwards WD, et al. Endomyocardial biopsy in 30 patients with primary amyloidosis and suspected cardiac involvement. Arch Intern Med 1988;148:662-6.
10. Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 2 of 2-diagnostic criteria and appropriate utilization. J Card Fail 2019;25:854-65.
11. Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016;133:2404-12.
12. Hamer JP, Janssen S, van Rijswijk MH, Lie KI. Amyloid cardiomyopathy in systemic non-hereditary amyloidosis. Clinical, echocardiographic and electrocardiographic findings in 30 patients with AA and 24 patients with AL amyloidosis. Eur Heart J 1992;13:623-7.
13. Quarta CC, Solomon SD, Uraizee I, et al. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation 2014;129:1840-9.
14. Phelan D, Collier P, Thavendiranathan P, et al. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart 2012;98:1442-8.
15. Williams LK, Forero JF, Popovic ZB, et al. Patterns of CMR measured longitudinal strain and its association with late gadolinium enhancement in patients with cardiac amyloidosis and its mimics. J Cardiovasc Magn Reson 2017;19:61.
16. Löfbacka V, Axelsson J, Pilebro B, Suhr OB, Lindqvist P, Sundström T. Cardiac transthyretin amyloidosis 99mTc-DPD SPECT correlates with strain echocardiography and biomarkers. Eur J Nucl Med Mol Imaging 2021;48:1822-32.
17. Steen H, Giusca S, Montenbruck M, et al. Left and right ventricular strain using fast strain-encoded cardiovascular magnetic resonance for the diagnostic classification of patients with chronic non-ischemic heart failure due to dilated, hypertrophic cardiomyopathy or cardiac amyloidosis. J Cardiovasc Magn Reson 2021;23:45.
18. Giusca S, Steen H, Montenbruck M, et al. Multi-parametric assessment of left ventricular hypertrophy using late gadolinium enhancement, T1 mapping and strain-encoded cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2021;23:92.
19. Maceira AM, Joshi J, Prasad SK, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2005;111:186-93.
20. Duca F, Kammerlander AA, Panzenböck A, et al. Cardiac magnetic resonance T1 mapping in cardiac amyloidosis. JACC Cardiovasc Imaging 2018;11:1924-6.
21. Burrage MK, Ferreira VM. Cardiovascular magnetic resonance for the differentiation of left ventricular hypertrophy. Curr Heart Fail Rep 2020;17:192-204.
22. Fontana M, Pica S, Reant P, et al. Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2015;132:1570-9.
23. Zhao L, Tian Z, Fang Q. Diagnostic accuracy of cardiovascular magnetic resonance for patients with suspected cardiac amyloidosis: a systematic review and meta-analysis. BMC Cardiovasc Disord 2016;16:129.
24. Syed IS, Glockner JF, Feng D, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging 2010;3:155-64.
25. Martinez-Naharro A, Treibel TA, Abdel-Gadir A, et al. Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol 2017;70:466-77.
26. Sado DM, White SK, Piechnik SK, et al. Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ Cardiovasc Imaging 2013;6:392-8.
27. Sado DM, Flett AS, Banypersad SM, et al. Cardiovascular magnetic resonance measurement of myocardial extracellular volume in health and disease. Heart 2012;98:1436-41.
28. Karamitsos TD, Piechnik SK, Banypersad SM, et al. Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging 2013;6:488-97.
29. Fontana M, Banypersad SM, Treibel TA, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging 2014;7:157-65.
30. Baggiano A, Boldrini M, Martinez-Naharro A, et al. Noncontrast magnetic resonance for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging 2020;13:69-80.
31. Banypersad SM, Sado DM, Flett AS, et al. Quantification of myocardial extracellular volume fraction in systemic AL amyloidosis: an equilibrium contrast cardiovascular magnetic resonance study. Circ Cardiovasc Imaging 2013;6:34-9.
32. Martinez-Naharro A, Kotecha T, Norrington K, et al. Native T1 and extracellular volume in transthyretin amyloidosis. JACC Cardiovasc Imaging 2019;12:810-9.
33. Pan JA, Kerwin MJ, Salerno M. Native T1 mapping, extracellular volume mapping, and late gadolinium enhancement in cardiac amyloidosis: a meta-analysis. JACC Cardiovasc Imaging 2020;13:1299-310.
34. Bandula S, Banypersad SM, Sado D, et al. Measurement of Tissue interstitial volume in healthy patients and those with amyloidosis with equilibrium contrast-enhanced MR imaging. Radiology 2013;268:858-64.
35. Fontana M, Banypersad SM, Treibel TA, et al. AL and ATTR cardiac amyloid are different: native T1 mapping and ECV detect different biology. J Cardiovasc Magn Reson 2014:16.
36. Fontana M, Banypersad SM, Treibel TA, et al. Differential myocyte responses in patients with cardiac transthyretin amyloidosis and light-chain amyloidosis: a cardiac MR imaging study. Radiology 2015;277:388-97.
37. Kotecha T, Martinez-Naharro A, Treibel TA, et al. Myocardial edema and prognosis in amyloidosis. J Am Coll Cardiol 2018;71:2919-31.
38. Sharpley FA, Fontana M, Martinez-Naharro A, et al. Cardiac biomarkers are prognostic in systemic light chain amyloidosis with no cardiac involvement by standard criteria. Haematologica 2020;105:1405-13.
39. Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis. A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur J Heart Fail 2021;23:512-26.
40. Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol 2005;79:319-28.
41. Grogan M, Scott CG, Kyle RA, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol 2016;68:1014-20.
42. Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2018;39:2799-806.
43. Banypersad SM, Fontana M, Maestrini V, et al. T1 mapping and survival in systemic light-chain amyloidosis. Eur Heart J 2015;36:244-51.
44. Messroghli DR, Moon JC, Ferreira VM, et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: a consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J Cardiovasc Magn Reson 2017;19:75.
45. Law S, Petrie A, Chacko L, et al. Disease progression in cardiac transthyretin amyloidosis is indicated by serial calculation of National Amyloidosis Centre transthyretin amyloidosis stage. ESC Heart Fail 2020;7:3942-9.
46. Raina S, Lensing SY, Nairooz RS, et al. Prognostic value of late gadolinium enhancement CMR in systemic amyloidosis. JACC Cardiovasc Imaging 2016;9:1267-77.
47. Boynton SJ, Geske JB, Dispenzieri A, et al. LGE provides incremental prognostic information over serum biomarkers in AL cardiac amyloidosis. JACC Cardiovasc Imaging 2016;9:680-6.
48. Lin L, Li X, Feng J, et al. The prognostic value of T1 mapping and late gadolinium enhancement cardiovascular magnetic resonance imaging in patients with light chain amyloidosis. J Cardiovasc Magn Reson 2018;20:2.
49. Dorbala S, Vangala D, Bruyere J Jr, et al. Coronary microvascular dysfunction is related to abnormalities in myocardial structure and function in cardiac amyloidosis. JACC Heart Fail 2014;2:358-67.
50. Chacko L, Kotecha T, Martinez-naharro A, et al. 1171Myocardial perfusion mapping in cardiac amyloidosis - exploring the spectrum from infiltration to ischaemia. Eur Heart J 2019;40:ehz748.0013.
52. Addison D, Slivnick JA, Campbell CM, Vallakati A, Jneid H, Schelbert E. Recent advances and current dilemmas in the diagnosis and management of transthyretin cardiac amyloidosis. J Am Heart Assoc 2021;10:e019840.
53. Martinez-Naharro A, Abdel-Gadir A, Treibel TA, et al. CMR-verified regression of cardiac AL amyloid after chemotherapy. JACC Cardiovasc Imaging 2018;11:152-4.
54. Maurer MS, Schwartz JH, Gundapaneni B, et al. ATTR-ACT Study Investigators. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018;379:1007-16.
55. Solomon SD, Adams D, Kristen A, et al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation 2019;139:431-43.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Pica S, Lombardi M. Disclosing the cardiomyopathic substrate within the heart muscles in amyloidosis by cardiac magnetic resonance: diagnostic and prognostic implications. Vessel Plus 2022;6:10. http://dx.doi.org/10.20517/2574-1209.2021.81
AMA Style
Pica S, Lombardi M. Disclosing the cardiomyopathic substrate within the heart muscles in amyloidosis by cardiac magnetic resonance: diagnostic and prognostic implications. Vessel Plus. 2022; 6: 10. http://dx.doi.org/10.20517/2574-1209.2021.81
Chicago/Turabian Style
Pica, Silvia, Massimo Lombardi. 2022. "Disclosing the cardiomyopathic substrate within the heart muscles in amyloidosis by cardiac magnetic resonance: diagnostic and prognostic implications" Vessel Plus. 6: 10. http://dx.doi.org/10.20517/2574-1209.2021.81
ACS Style
Pica, S.; Lombardi M. Disclosing the cardiomyopathic substrate within the heart muscles in amyloidosis by cardiac magnetic resonance: diagnostic and prognostic implications. Vessel Plus. 2022, 6, 10. http://dx.doi.org/10.20517/2574-1209.2021.81
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.