
Molecular genetic analysis of cerebral cavernous malformations: an update

Abstract
Cerebral cavernous malformations (CCM) can occur either as sporadic or familial form with autosomal dominant inheritance. Three CCM genes have been identified: CCM1 (KRIT1), CCM2 (MGC4607), and CCM3 (PDCD10). In this review, we provide an overall update on genetics of cerebral cavernous malformations. We discuss the main features of these three genes and provide an updated listing of the mutations identified so far. Most of them lead to a premature stop codon regardless of the nature of the variation, including nonsense mutations, small deletions/insertions, and intronic/exonic substitutions causing an altered splicing and a frame-shift. In addition, deletions or duplications of one or more exons of CCM genes can be responsible for the disease. We examine the use of different mutation screening methods to identify all these mutations, providing a comprehensive approach to CCM genetic diagnosis. We also report the main strategies to evaluate the actual impact of the mutations on the protein function. Moreover, we recapitulate the available data on penetrance, phenotype-genotype correlations, and founder effect. Finally, we discuss the main aspects of genetic counseling, including genetic risk assessment in family members, in sporadic patients with multiple CCMs, and in the case of de novo mutations.
Keywords
INTRODUCTION
Cerebral cavernous malformations (CCM) are vascular lesions that can occur as a sporadic (80% of cases) or a familial autosomal dominant disorder (FCCM) (20% of cases), with incomplete clinical and neuroradiological penetrance and great inter-individual variability[1].
Sporadic forms usually present with a single lesion on MRI[2,3], although multiple lesions have been reported in some cases[4,5]. In contrast, familial forms typically exhibit multiple lesions, which increase in number and size over time[6]. CCM result in a variety of clinical manifestations, including recurrent headaches, seizures, focal neurological deficit, and hemorrhage, with onset usually during adult life, but symptoms can also start in early infancy or in old age[1].
In addition to neural lesions, extraneural cavernous malformations have been described in the familial form, in particular cutaneous and retinal vascular malformations[7,8].
Three CCM genes have been identified so far: CCM1/KRIT1[9], CCM2/MGC4607[10], and CCM3/PDCD10[11]. Almost 80% of CCM patients affected with a genetic form of the disease carry a heterozygous germline mutation in one of these genes[1].
The purpose of this review is to provide an update on recent advances in molecular genetics of cerebral cavernous malformations. We provide the main features of the three genes and an updated listing of CCM pathogenic variants published so far in the peer-reviewed literature. We summarize the available data on penetrance, phenotype-genotype correlations, and founder effect for the variants described in the three CCM genes. Moreover, we briefly examine the different mutation screening methods in the genetic diagnostic approach to CCM and discuss the main aspects of genetic counseling.
CCM GENES
Mutations in the KRIT1 gene have been found in 53%-65% of the familial forms of CCM. Mutations in the CCM2 gene account for approximately 20% of the familial CCM cases, whereas 10%-16% of CCM families harbor mutations in the PDCD10 gene[12].The existence of a potential fourth gene linked to CCM has long been postulated, since 5%-15% of familial cases cannot be explained by mutations in the three known CCM genes. It is however rather unlikely now, more than 15 years after the identification of the PDCD10 gene[11]. It is probable that the very few CCM families apparently negative for mutations in KRIT1, CCM2, and PDCD10 genes harbor a pathogenic variant not identified by the routinely used techniques (e.g., a variant outside the screened exonic regions or a copy number neutral genomic rearrangement in one of the three genes[13]).
Thus far, more than 350 different KRIT1/CCM2/PDCD10 germline mutations have been identified and included in the Human Gene Mutation Database (HGMD)[14]. These variants are highly stereotyped. Almost all of them introduce a premature termination codon in the protein, through various mechanisms, such as nonsense, splice-site, and frameshift mutations, as well as larger genomic rearrangements. Of note, even though the consequences are highly stereotyped, germline CCM mutations are usually present in only one or few families and are rarely recurrent[1].
The molecular mechanisms responsible for the formation of CCM lesions in the presence of CCM gene mutations remain unclear. The proteins encoded by these genes are essential for regulating the angiogenesis during embryonic development and vascular postnatal stages of development[9,15-17]. They are involved in the maintenance of junctional integrity between adjacent vascular endothelial cells[18,19] and are expressed in endothelium, neurons, and astrocytes[20,21]. Endothelial cells seem to be the cell of origin for CCM[22,23].
CCM1/KRIT1
The KRIT1 gene (OMIM #604214) is located on chromosome 7q21.2 and contains 16 coding exons[24] that encode for the Krev interaction trapped 1 (KRIT1) protein (UniProt #O00522). KRIT1 is a 736-amino acid microtubule-associated protein, containing a NUDIX domain, multiple NPxY/F motifs, four ankyrin repeat domains at the N-terminus, and one C-terminal FERM domain (band 4.1 ezrin radixin moesin domain)[25,26]. Gene and protein structures are shown in Figure 1.
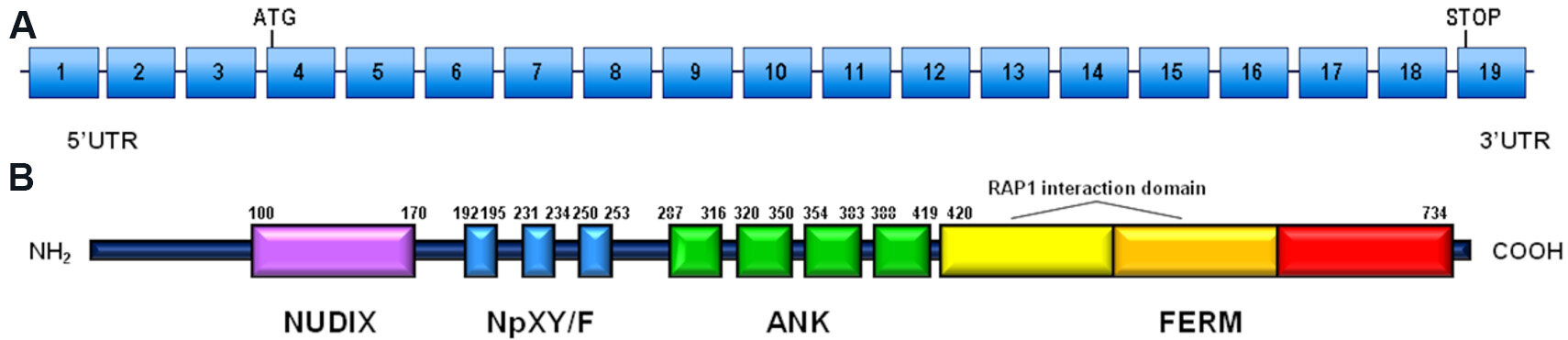
Figure 1. Schematic representation of KRIT1 gene and KRIT1 protein: (A) KRIT1 gene structure including 19 coding exons (rectangles) and introns (horizontal lines) (reference sequence: NM_004912.3); and (B) KRIT1 protein structure, including NUDIX domain, NPxY/F motifs, ankyrin repeat domains (ANK), and FERM domain (band 4.1 ezrin radixin moesin domain), composed of three subdomains. The position of the amino acid at the beginning and at the end is indicated for each domain.
The NPxY/F motifs may be involved in dimerization and intramolecular folding of the KRIT1 protein[26] and are recognized by phosphotyrosine binding (PTB) domains. PTB domains are present on several proteins, including the α-isoform of β1-integrin regulator integrin cytoplasmic adaptor protein 1 (ICAP1α)[27]. Through the same PTB domain, ICAP1α can interact with KRIT1 or with integrins. In the latter case, the interaction causes the activation of β-integrin signaling and stimulates angiogenesis. Thus, KRIT1 may act as a modulator of ICAP1α activity, by competing with β-integrin interaction[28]. The NpxY/F motifs also interact with other known proteins: sorting nexin 17 (SNX17), a modulator of endocytosis and intracellular trafficking[29]; the Kelch family protein Nd1-L, showing a role in ROS (Reactive Oxigen Species) homeostasis[30]; and the PTB domain present in Malcavernin, which in turn binds to PDCD10 and acts as a bridge between KRIT1 and PDCD10[26].
The ankyrin repeats in KRIT1, present in many proteins, mediate inter- and intra-molecular interactions. They are involved in different cellular processes, such as gene transcription, cell cycle control, and organization of the cytoskeleton[31]. Finally, the FERM domain of KRIT1, composed of three subdomains F1-F3, interacts with the NPxY/F motif on the cytoplasmic face of transmembrane receptors[24] and with Rap1. Association with Rap1 relocalizes KRIT1 from microtubules to cell junction membranes[32].
KRIT1 pathogenic variants
More than 300 pathogenic variants have been reported so far in the HGMD[14]. These variants are present across the whole gene, with no evidence of hot spot regions [Supplementary Table 1]. The majority of them are substitutions, deletions, and insertions [Figure 2A], mainly located in the coding and splicing regions [Figure 2B]. They are splice junctions, frameshift, nonsense, and missense variants, often affecting the splicing process [Figure 2C][12]. Gross deletions, involving one or more exons, until the complete lack of the gene, have also been reported.
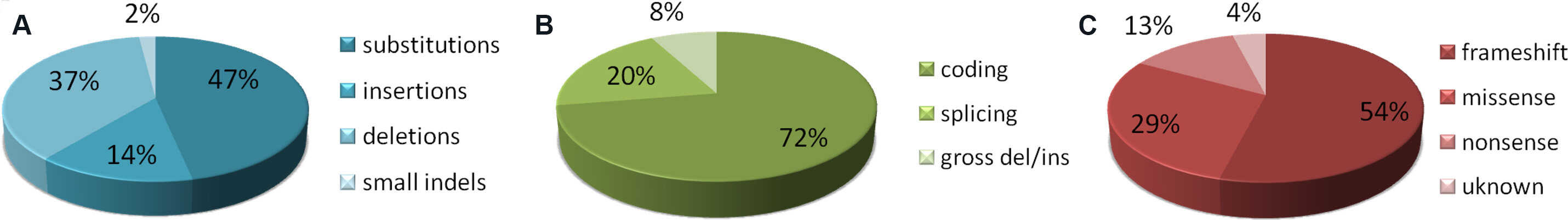
Figure 2. Mutation distribution of pathogenic KRIT1 variants, as reported so far in the HGMD[14]: (A) distribution by type at DNA level; (B) distribution by DNA location; and (C) distribution by type at protein level (please note that in this case the term “missense” is used to define variants so classified in the original studies, without information regarding their impact on splicing process). The classification of variants has not been independently verified by the authors.
Regardless of the type of mutation, pathogenic variants result in a premature termination of translation, introducing an early termination codon, generating an unstable mRNA, or truncated KRIT1 proteins totally or partially lacking in the putative Rap1-interacting region[33]. This evidence supports the hypothesis of a loss-of-function mechanism[34], which can lead to CCM lesion genesis. Loss of KRIT1 alters cellular signaling and behavior. Moreover, endothelial cells acquire stem cell-like features and become more proliferative and invasive[35].
CCM2/MGC4607
The CCM2/MGC4607 gene (OMIM #607929) is located on chromosome 7p13 and contains 10 coding exons[24]. CCM2 encodes for CCM2/Malcavernin (UniProt #Q9BSQ5), a 444-amino acid protein that contains a predicted N-terminal PTB domain and a C-terminal Harmonin homology domain (HHD) [Figure 3].
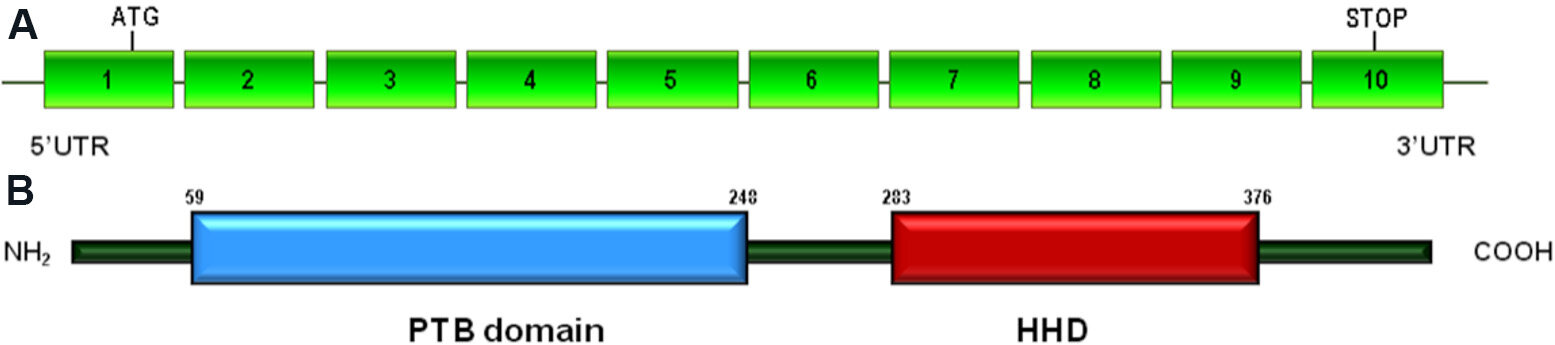
Figure 3. Schematic representation of CCM2 gene and Malcavernin protein: (A) CCM2 gene structure including 10 coding exons (rectangles) and introns (horizontal lines) (reference sequence: NM_031443.3); and (B) Malcavernin protein structure, including the N-terminal phosphotyrosine binding (PTB) domain and the C-terminal Harmonin homology domain (HHD). The position of the amino acid at the beginning and at the end is indicated for each domain.
Through the PTB domain, Malcavernin binds to KRIT1 and regulates its cellular localization[26,36]. The HHD domain is involved in the interaction with the protein kinase MEKK3 (MAP3K3)[37]. As a result of this interaction, Malcavernin acts as a scaffold protein in the signaling cascade that controls the activation of p38 MAPK[33]. A linker region, located between the PTB and HHD domains, binds to PDCD10 through its N-terminal portion[38].
Due to all these interactions, Malcavernin plays a pivotal role in signal transduction pathways that regulate adhesion, cytoskeleton remodeling, proliferation, migration of cells, and, ultimately, maintenance of vascular integrity[33].
CCM2 pathogenic variants
To date, more than 90 mutations in the CCM2 gene have been identified and listed in HGMD
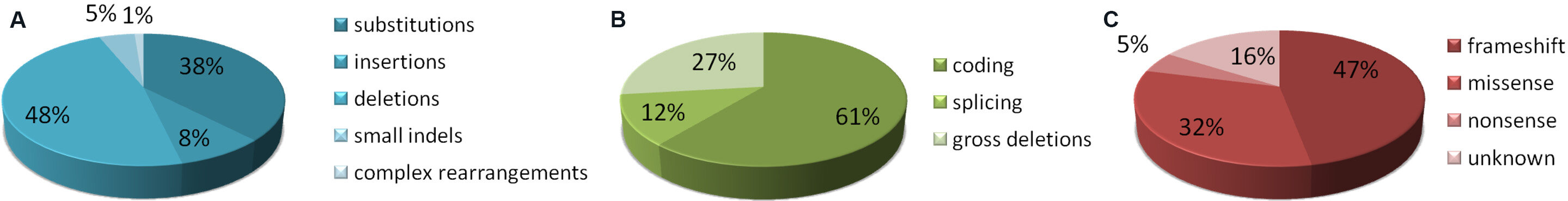
Figure 4. Mutation distribution of pathogenic CCM2 variants as reported so far in the HGMD[14]: (A) distribution by type at DNA level; (B) distribution by DNA location; and (C) distribution by type at protein level (please note that in this case the term “missense” is used to define variants so classified in the original studies, without information regarding their impact on splicing process). The classification of variants has not been independently verified by the authors.
As in the case of the KRIT1 gene, deletions involving whole gene or a large part of it have been described. A 77.6-kb deletion spanning from exon 2 to exon 10 is a common founder mutation in the United States population[42].
CCM3/PDCD10
The PDCD10/CCM3 gene (OMIM #609118), located on chromosome 3q26.1, is a highly conserved gene containing seven coding and three non-coding exons[24]. It encodes the programmed cell death protein 10 (PDCD10), a 212-amino acid protein (UniProt #Q9BUL8) that contains a dimerization domain at the N-terminus, and a C-terminal focal adhesion targeting (FAT)-homology domain [Figure 5].
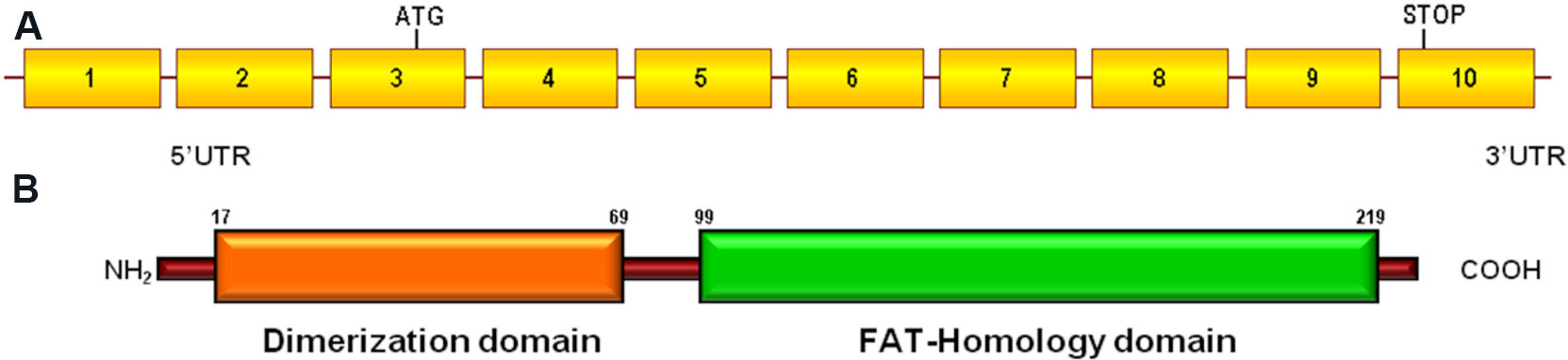
Figure 5. Schematic representation of PDCD10 gene and PDCD10 protein: (A) PDCD10 gene structure including 10 exons (rectangles) and introns (horizontal lines) (reference sequence: NM_007217.3); and (B) PDCD10 protein structure, including the N-terminal dimerization domain and the C-terminal focal adhesion targeting (FAT)-homology domain. The position of the amino acid at the beginning and at the end is indicated for each domain.
The dimerization domain allows PDCD10 to form a dimer. The FAT-homology domain stabilizes the PDCD10 protein and interacts with Malcavernin[38] and with several other signaling proteins[43,44]. Among these are molecules involved in VEGF signaling, which is fundamental for vascular development[45,46]. In addition, PDCD10 plays a critical role in the regulation of the angiogenesis through DLL4-Notch signaling[45,47].
It is known that PDCD10 is involved in apoptosis: its overexpression induces the activation of caspase 3 and increases cell death[48,49]. Thus, in the presence of PDCD10 mutations, CCM disease may originate from a modification of the apoptotic process, which alters the equilibrium between neural cells and endothelium[50].
PDCD10 pathogenic variants
More than 70 pathogenic variants of the PDCD10 gene are present in the HGMD [Supplementary Table 3], including small and gross deletions, substitutions, insertions, and duplications [Figure 6A]. They are mainly located in the coding region, often between exon 5 and exon 7. In this case as well, frameshift, nonsense, and missense variants have been identified, all leading to a premature stop codon. Moreover, the presence of large deletions may result in the lack of protein production [Figure 6B and C].
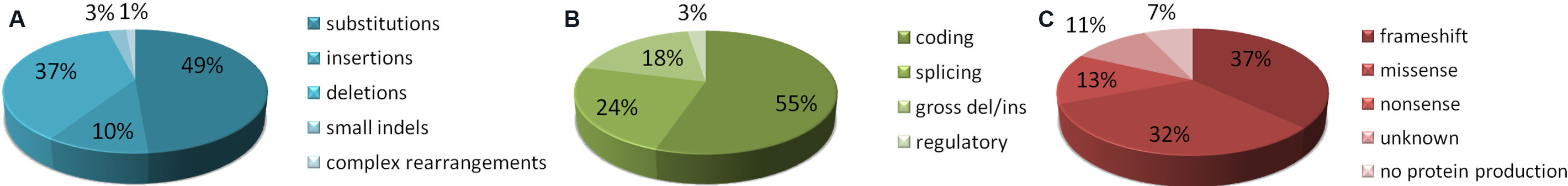
Figure 6. Mutation distribution of pathogenic PDCD10 variants as reported so far in the HGMD[14]: (A) distribution by type at DNA level; (B) distribution by DNA location; and (C) distribution by type at protein level (please note that in this case the term “missense” is used to define variants so classified in the original studies, without information regarding their impact on splicing process). The classification of variants has not been independently verified by the authors.
Considering that the pathogenic variants detected so far are loss of function variants, haploinsufficiency and somatic loss of heterozygosity have been proposed as pathogenic mechanisms for CCM3[23,51].
PHENOTYPE CORRELATIONS BY GENE
An important feature of CCM is the heterogeneity of the phenotypes. Variable expressivity is observed within affected individuals belonging to the same family and between families linked to the same mutation. To date, only few data are available on genotype-phenotype correlation. Several studies have established some potential phenotype correlations by gene in FCCM.
Familial cases with KRIT1 mutations have been demonstrated to have less severe clinical manifestations than the other familial forms[52]. Up to 60% of patients with FCCM caused by a pathogenic variant in KRIT1 gene ultimately become symptomatic[53]. Moreover, cutaneous vascular malformations have been identified in 9% of FCCM patients. They have been reported more commonly in familial patients with KRIT1 gene variants[7] than in patients with pathogenic variant in CCM2 or PDCD10. Three distinct major cutaneous vascular malformation phenotypes have been identified, namely hyperkeratotic cutaneous capillary-venous malformation (HCCVM), capillary malformation (CM), and venous malformation (VM), accounting for 39%, 34%, and 21% of FCCM cases, respectively[7].
In contrast to most patients with KRIT1 and CCM2 genotypes, who often live normal lives with rarely disabling clinical manifestations, patients with PDCD10 mutations appear to be related to more specific and severe phenotype. They have greater lesion burden, a higher risk for cerebral hemorrhage, and an earlier onset before the age of 15 years[54-56]. The difference in hemorrhage risk, compared to patients with KRIT1 and CCM2 mutations, seems apparently not related to the size or number of lesions.
There is also a significant association with other clinical features, which include skin lesions, scoliosis, spinal cord cavernous malformations, brain tumor (meningioma, astrocytoma, and acoustic neuroma), and cognitive disability, unrelated to lesion burden or hemorrhage. Interestingly, an association with skin lesions had been reported primarily in familial patients with KRIT1 variants. However, in patients with PDCD10 mutations, lesions are different (more café-au-lait lesions rather than hyperkeratotic angiomas)[56]. In addition, PDCD10 mutation carriers have a greater likelihood of de novo mutational event than KRIT1 and CCM2 mutation carriers[13].
Comparison of the MRI features of KRIT1, CCM2, and PDCD10 mutation carriers suggested that the increment with age of the number of lesions on MRI gradient echo sequence (GRE) varies according to the mutated CCM gene. Fewer brain lesions on GRE MRI and a slower rate of lesion development have been observed in CCM2 mutation carriers compared to patients with KRIT1 gene variants[54].
PENETRANCE
Clinical penetrance varies among the CCM genes and may be specific to the pathogenic variant[57]. Clinical penetrance is estimated to be 60%-88% in CCM1, 100% in CCM2, and 63% in CCM3 families[53,58,59]. Within KRIT1 positive families, both clinical and neuroradiological penetrance are incomplete and age dependent. Neuroradiological penetrance of CCM was previously considered to be complete or almost complete. Nevertheless, molecular screening of asymptomatic individuals revealed that cerebral MRI penetrance is also incomplete, even using the highly sensitive GRE sequences, and age dependent[53,60].
Among the three CCM genes, CCM2 is the only one reported with 100% of clinical and neuroradiological penetrance[59]. However, in 2018, Scimone et al.[61] found seven members of a family with a novel mutation, c.1555-1G>A (published as IVS10-1G>A), in whom only five of them showed lesions on MRI scan. Therefore, for the first time, a penetrance < 100%, equivalent to 70%, was reported for the CCM2 gene.
FOUNDER PATHOGENIC VARIANTS
To date, more than 350 different mutations in the three CCM genes have been reported in the HGMD. Although most of them are family specific, four founder pathogenic variants have been identified so far that may be useful for stratifying genetic analysis in specific populations.
A founder effect has been shown for the pathogenic variant c.1363C>T (p.Gln455Ter) in the KRIT1 gene, referred to as “the common Hispanic variant”, identified in about 70% of affected families with ancestry from north Mexico and the American Southwest[62,63].
A 77.6-kb deletion spanning exons 2-10 of the CCM2 gene resulted a common founder deletion with a high prevalence (up to 22%) in the United States population[42,64], while it was rare in the Italian population[65].
In the Italian population, a founder effect in four Sardinian families was observed for the mutation c.987C>A (p.Cys329Ter) in exon 10 of the KRIT1 gene[66].
More recently, a two-base pair change, c.30+5_30+6delinsTT, affecting messenger RNA splicing of the CCM2 gene, was identified in seven apparently unrelated probands from 10 different kindreds of Ashkenazi Jewish descent, resulting a founder mutation in the Ashkenazi Jewish population[67].
Finally, a redundant 14-bp deletion (c.554_567del) in exon 5 of CCM2 gene has been described in patients from the Iberian Peninsula[68,69]. However, in this case, a haplotype study has not been performed to determine a founder effect.
The main features of the founder pathogenic variants are summarized in Table 1.
Founder pathogenic variants identified so far
| Gene | DNA nucleotide change | Predicted protein change | Reference sequences | Population | Ref. |
| KRIT1 | c.987C>A | p.Cys329Ter | NM_194456.1 NP_919438.1 | Sardinian | [62] |
| KRIT1 | c.1363C>T | p.Gln455Ter | North Mexican and American Southwest ancestry | [58,59] | |
| CCM2 | c.30+5_30+6delGCinsTT | -- | NM_031443.3 NP_113631.1 | Ashkenazi Jewish | [63] |
| CCM2 | 77.6-kb deletion (exons 2–10) | -- | United States | [38,60] |
GENETIC TESTING
The molecular testing approach for CCM includes several methods to ensure the best diagnostic accuracy. A stepwise protocol may provide a high screening sensitivity, rationalizing costs and times of the analysis.
The first step is usually the sequencing of all coding exons and exon-intron boundaries of the three CCM genes, on genomic DNA obtained from blood cells. A serial single gene testing, using Sanger sequencing, or a multigene panel in Next Generation Sequencing (NGS) can be performed. Both techniques allow the identification of point mutations with a very high sensitivity. NGS is a faster and more time-effective method, since it allows analyzing all genes in the same session. However, sequence variants identified by NGS still have to be confirmed by Sanger sequencing.
When no mutation is identified by sequencing, the next step is a quantitative analysis of exon copy number, to evaluate the presence of deletions or duplications affecting one or multiple exons in the three CCM genes. This analysis can be performed by several approaches: Quantitative Multiplex PCR Short Fragments (QMPSF), gene-targeted microarray designed to detect exon deletions or duplications, or Multiplex Ligation-dependent Probe Amplification (MLPA). The last method is in general less expensive; ensures high reproducibility, since it is supplied as a tested and proven commercial kit; and shows a very low coefficient of variation when compared to quantitative PCR[70]. NGS can also be used to identify copy number variations with algorithms based on sequencing reads depth analysis. However, at the moment, this approach is less sensitive and less specific than MLPA, which remains the best solution for detecting exon deletion/duplication[71].
If a variant is identified in the first two steps, its role has to be evaluated. If the variant introduces a premature stop codon (nonsense or frameshift variant, caused by point mutations or exon deletions) or is located in an invariant splicing region, its pathogenic role is easy to define. The pathogenic features of missense mutations are more difficult to establish. Indeed, it has been demonstrated that some “missense” mutations actually activate cryptic splice sites and cause an aberrant splicing of CCM mRNA, resulting in frameshift and introduction of a premature stop codon[1,72,73]. In these cases, a predictive in silico analysis, using tools such as NetGene2 server (http://www.cbs.dtu.dk/services/NetGene2)[74], may be useful to evaluate the impact of the variant on mRNA. However, an analysis at the cDNA level is always highly recommended to verify the presence of aberrant splicing and correctly define the role of the variants. The same approach is suggested also in the case of variants located in the splicing regions. This can be easily performed, since mRNA can be extracted from peripheral leukocytes, where CCM genes are expressed, reverse transcribed into cDNA, and analyzed by PCR.
In rare cases, some missense variants may impair the interactions between KRIT1 and CCM2 and undermine the stability of the CCM complex[39]. It has been demonstrated that some mutations located in the PTB domain of CCM2 are able to destroy the interaction with KRIT1[38,40,41]. Thus, missense variants affecting PTB domain of CCM2 may be considered pathogenic, and their role should be verified by performing functional studies, to evaluate whether they damage the CCM2-KRIT1 interaction, as previously described[39].
The genomic DNA sequencing followed by copy number analysis allows identifying pathogenic variants in 87%-98% of all familial CCM cases[13]. In a minority of patients with a positive family history of CCM or with multiple CCM lesions, however, this approach does not allow detecting any mutations. For these cases, pathogenic variants outside of the standard diagnostic target regions may be considered. In addition, in that event, cDNA analysis is advisable. It can reveal splicing anomalies produced by deep intronic variants, not revealed by standard sequencing, or a loss of heterozygosity causing the lack of expression of one of the CCM alleles. Some CCM deep intronic variants have been identified by using this approach. An example is a deep intronic KRIT1 gene deletion (c.262+132_262+133delAA), which leads to the insertion of a 99-bp pseudo-exon causing a premature stop codon in the open reading frame[75].
Finally, rare structural anomalies, such as inversions, can be detected using whole genome sequencing (WGS) and bioinformatics analysis. An example is the recent identification of a 24-kb inversion involving exon 1 of CCM2 gene[76]. However, this kind of approach is currently performed in a research context and is not part of the standard diagnostic process.
A flow-chart summarizing the approach to CCM genetic screening is shown in Figure 7.
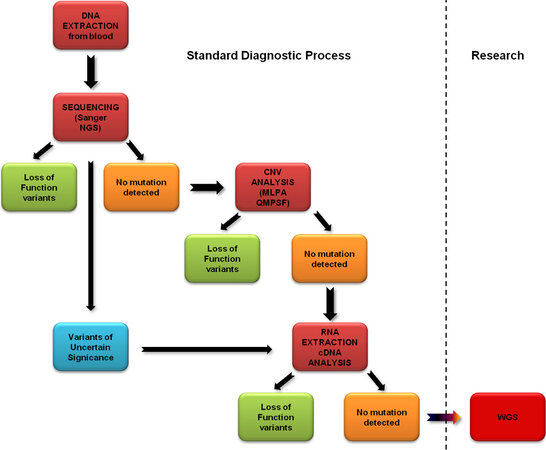
Figure 7. Stepwise protocol of CCM gene screening in familial CCM cases (probands) and sporadic CCM cases with multiple lesions. After genomic DNA extraction from white blood cells, sequencing of all exons and intron-exon boundaries of the three CCM genes is performed, using serial single gene testing by Sanger sequencing or multigene panels by Next Generation Sequencing (NGS). If no mutation is detected, the analysis is extended to exon copy number variations (CNV) using quantitative MLPA or QMPSF. If negative, RNA extracted from blood leukocytes is reverse transcribed and sequenced by Sanger sequencing to evaluate the presence of aberrant splicing. This approach is also used in the case of variants of uncertain significance identified in the first step of the analysis. These approaches are routinely performed in diagnostic labs. These steps are part of the standard diagnostic process. If no alteration is detected, whole genome sequencing (WGS) may be performed to assess the presence of rare structural anomalies (usually in a research context). CCM: Cerebral cavernous malformations.
GENETIC COUNSELING
Genetic testing for KRIT1, CCM2, and PDCD10 genes can confirm the clinical diagnosis in patients and guide the genetic counseling. However, to correctly evaluate the genetic risk of CCM, it is necessary to assess whether the patient has a family history of disease and presents with single or multiple CCM lesions, in the absence of developmental venous anomaly or history of brain radiation. Thus, a detailed three-generation pedigree providing information about probable CCM symptoms, such as seizures, recurrent headaches, cerebral hemorrhages, focal neurological deficits, and a proband’s brain MRI, including gradient echo (GRE) or susceptibility-weighted imaging (SWI) sequences, should be obtained.
It is necessary to take into account that the family history can be negative because the disease is not recognized in other family members. This could be due to parents’ death before symptom onset, reduced penetrance, or phenotypic variability. Thus, in the presence of an apparently negative family history, it is still advisable to perform the appropriate evaluations in the proband’s parents.
In the case of familial forms of CCM, the genetic screening sensitivity for the three CCM genes in a proband with an affected relative is more than 90%. When the proband carries a mutation, the sensitivity of screening in his/her relatives reaches 100%[77]. If a mutation is identified in a proband, genetic testing of at-risk family members can be offered. Genetic counseling is fundamental to give patients and relatives all details necessary to make an informed choice. In particular, asymptomatic individuals should be carefully informed about the possible psychological implications of a positive test before they take a decision. Moreover, proband’s family members should be informed that about 40% of the CCM mutation carriers remain asymptomatic[54] and intrafamilial phenotypic variability may be high. Predictive testing in minors raises ethical issues and should not be carried out[24].
In the case of sporadic forms of CCM, in individuals with a single lesion, the presence of CCM gene mutations is exceedingly rare. A study on KRIT1 gene mutations in sporadic cases showed the presence of mutations in 29% of sporadic cases with multiple lesions, while no mutation was identified in patients with single lesions[5]. For this reason, genetic testing is usually not indicated in sporadic forms with a single lesion.
On the other hand, sporadic cases with multiple CCM lesions likely carry a pathogenic variant and should be managed as familial cases. Thus, genetic screening of all three CCM genes is recommended. However, in these cases, the mutation detection rate is close to 60%, a value much lower than in familial cases[13,78]. The patient should be informed that, even in the presence of a negative test, a genetic cause cannot be ruled out. There can be several explanations for a negative test: it may be because somatic mosaicism of de novo mutations occurred during gestation and therefore are not detectable in DNA from peripheral leukocytes; mutations may not be detectable because they are located outside the regions usually analyzed by conventional methods (i.e., in regulatory regions far from coding exons); and epigenetic modifications able to alter the expression of CCM proteins may occur.
When a pathogenic variant is detected in the proband, the analysis may be extended to the parents, in the same way as described for familial cases. Data reported so far indeed show that most sporadic patients with multiple lesions are cases with mutations inherited from an asymptomatic parent. Alternatively, they may be affected by CCM as the result of a de novo mutation[1,77]. In the latter case, the mutation is not found in proband’s parents.
The real percentage of cases caused by de novo pathogenic variants is unknown, because genetic analysis in parents is not always possible[79]. Individuals carrying a de novo germline pathogenic variant in all three CCM genes, most frequently in PDCD10, have been reported[10,11,53,54,56,64,65,80]. These mutations may occur randomly at any stage of embryonic development or at the germline level in the parents’ gametes. However, other possible explanations, including incorrect attribution of paternity or maternity (in the case of assisted reproduction), could be considered. All these aspects should be considered for a correct genetic counseling.
CONCLUSIONS
In the last years, great progress has been obtained in understanding the genetic bases of CCM and in CCM patients’ characterization. The advances of the molecular biology technologies, as well as a combined use of different methods, may now unravel the genetic cause of the majority of CCM cases. Genetic data should always be associated with a detailed clinical characterization of patients and, when possible, their affected family members. This may allow better defining several aspects of the disease, such as the relationship between phenotypes and genotypes and the penetrance of the variants identified in the three CCM genes. In this respect, it is of fundamental importance for the scientific community to have access to updated databases reporting genetic and phenotypic data derived from the literature. This approach may allow reaching a deeper knowledge of the disease, to better understand CCM pathophysiology, guide genetic counseling, and eventually improve clinical care of patients.
DECLARATIONS
Authors’ contributions
Made substantial contributions to conception and design of the study: Battistini S, Ricci C
Performed graphical design, as well as provided technical, and material support: Riolo G
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2021.
REFERENCES
1. Riant F, Bergametti F, Ayrignac X, Boulday G, Tournier-Lasserve E. Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. FEBS J 2010;277:1070-5.
2. Labauge P, Laberge S, Brunereau L, Levy C, Tournier-lasserve E. Hereditary cerebral cavernous angiomas: clinical and genetic features in 57 French families. Lancet 1998;352:1892-7.
3. Petersen TA, Morrison LA, Schrader RM, Hart BL. Familial versus sporadic cavernous malformations: differences in developmental venous anomaly association and lesion phenotype. AJNR Am J Neuroradiol 2010;31:377-82.
4. Labauge P, Brunereau L, Coubes P, et al. Appearance of new lesions in two nonfamilial cerebral cavernoma patients. Eur Neurol 2001;45:83-8.
5. Verlaan DJ, Laurent SB, Sure U, et al. CCM1 mutation screen of sporadic cases with cerebral cavernous malformations. Neurology 2004;62:1213-5.
6. Rigamonti D, Hadley MN, Drayer BP, et al. Cerebral cavernous malformations. Incidence and familial occurrence. N Engl J Med 1988;319:343-7.
7. Sirvente J, Enjolras O, Wassef M, Tournier-Lasserve E, Labauge P. Frequency and phenotypes of cutaneous vascular malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. J Eur Acad Dermatol Venereol 2009;23:1066-72.
8. Labauge P, Krivosic V, Denier C, Tournier-Lasserve E, Gaudric A. Frequency of retinal cavernomas in 60 patients with familial cerebral cavernomas: a clinical and genetic study. Arch Ophthalmol 2006;124:885-6.
9. Laberge-le Couteulx S, Jung HH, Labauge P, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet 1999;23:189-93.
10. Denier C, Goutagny S, Labauge P, et al. Société Française de Neurochirurgie. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet 2004;74:326-37.
11. Bergametti F, Denier C, Labauge P, et al. Société Française de Neurochirurgie. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet 2005;76:42-51.
12. Morrison L, Akers A. Cerebral cavernous malformation, familial. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021.
13. Spiegler S, Rath M, Paperlein C, Felbor U. Cerebral cavernous malformations: an update on prevalence, molecular genetic analyses, and genetic counselling. Mol Syndromol 2018;9:60-9.
14. Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet 2020;139:1197-207.
15. Clatterbuck RE, Eberhart CG, Crain BJ, Rigamonti D. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J Neurol Neurosurg Psychiatry 2001;71:188-92.
16. Gunel M, Laurans MS, Shin D, et al. KRIT1, a gene mutated in cerebral cavernous malformation, encodes a microtubule-associated protein. Proc Natl Acad Sci U S A 2002;99:10677-82.
17. Whitehead KJ, Plummer NW, Adams JA, Marchuk DA, Li DY. Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development 2004;131:1437-48.
18. Yadla S, Jabbour PM, Shenkar R, Shi C, Campbell PG, Awad IA. Cerebral cavernous malformations as a disease of vascular permeability: from bench to bedside with caution. Neurosurg Focus 2010;29:E4.
19. McDonald DA, Shi C, Shenkar R, et al. Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum Mol Genet 2014;23:4357-70.
20. Seker A, Pricola KL, Guclu B, Ozturk AK, Louvi A, Gunel M. CCM2 expression parallels that of CCM1. Stroke 2006;37:518-23.
21. Stockton RA, Shenkar R, Awad IA, Ginsberg MH. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med 2010;207:881-96.
22. Glading A, Han J, Stockton RA, Ginsberg MH. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol 2007;179:247-54.
23. Pagenstecher A, Stahl S, Sure U, Felbor U. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet 2009;18:911-8.
24. Haasdijk RA, Cheng C, Maat-Kievit AJ, Duckers HJ. Cerebral cavernous malformations: from molecular pathogenesis to genetic counselling and clinical management. Eur J Hum Genet 2012;20:134-40.
25. Marchuk DA, Srinivasan S, Squire TL, Zawistowski JS. Vascular morphogenesis: tales of two syndromes. Hum Mol Genet 2003;12 Spec No 1:R97-112.
26. Faurobert E, Albiges-Rizo C. Recent insights into cerebral cavernous malformations: a complex jigsaw puzzle under construction. FEBS J 2010;277:1084-96.
27. Zawistowski JS, Serebriiskii IG, Lee MF, Golemis EA, Marchuk DA. KRIT1 association with the integrin-binding protein ICAP-1: a new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum Mol Genet 2002;11:389-96.
28. Scimone C, Donato L, Marino S, Alafaci C, D'Angelo R, Sidoti A. Vis-à-vis: a focus on genetic features of cerebral cavernous malformations and brain arteriovenous malformations pathogenesis. Neurol Sci 2019;40:243-51.
29. Czubayko M, Knauth P, Schlüter T, Florian V, Bohnensack R. Sorting nexin 17, a non-self-assembling and a PtdIns(3)P high class affinity protein, interacts with the cerebral cavernous malformation related protein KRIT1. Biochem Biophys Res Commun 2006;345:1264-72.
30. Guazzi P, Goitre L, Ferro E, et al. Identification of the Kelch family protein Nd1-L as a novel molecular interactor of KRIT1. PLoS One 2012;7:e44705.
31. Sedgwick SG, Smerdon SJ. The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem Sci 1999;24:311-6.
32. Béraud-Dufour S, Gautier R, Albiges-Rizo C, Chardin P, Faurobert E. Krit 1 interactions with microtubules and membranes are regulated by Rap1 and integrin cytoplasmic domain associated protein-1. FEBS J 2007;274:5518-32.
33. Cavalcanti DD, Kalani MY, Martirosyan NL, Eales J, Spetzler RF, Preul MC. Cerebral cavernous malformations: from genes to proteins to disease. J Neurosurg 2012;116:122-32.
34. Verlaan DJ, Davenport WJ, Stefan H, Sure U, Siegel AM, Rouleau GA. Cerebral cavernous malformations: mutations in Krit1. Neurology 2002;58:853-7.
35. Maddaluno L, Rudini N, Cuttano R, et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013;498:492-6.
36. Liquori CL, Berg MJ, Siegel AM, et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet 2003;73:1459-64.
37. Fisher OS, Deng H, Liu D, et al. Structure and vascular function of MEKK3-cerebral cavernous malformations 2 complex. Nat Commun 2015;6:7937.
38. Draheim KM, Li X, Zhang R, et al. CCM2-CCM3 interaction stabilizes their protein expression and permits endothelial network formation. J Cell Biol 2015;208:987-1001.
39. Bergametti F, Viot G, Verny C, et al. Novel CCM2 missense variants abrogating the CCM1-CCM2 interaction cause cerebral cavernous malformations. J Med Genet 2020;57:400-4.
40. Zawistowski JS, Stalheim L, Uhlik MT, et al. CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet 2005;14:2521-31.
41. Fisher OS, Liu W, Zhang R, et al. Structural basis for the disruption of the cerebral cavernous malformations 2 (CCM2) interaction with Krev interaction trapped 1 (KRIT1) by disease-associated mutations. J Biol Chem 2015;290:2842-53.
42. Liquori CL, Berg MJ, Squitieri F, et al. Deletions in CCM2 are a common cause of cerebral cavernous malformations. Am J Hum Genet 2007;80:69-75.
43. Hilder TL, Malone MH, Bencharit S, et al. Proteomic identification of the cerebral cavernous malformation signaling complex. J Proteome Res 2007;6:4343-55.
44. Li X, Zhang R, Zhang H, et al. Crystal structure of CCM3, a cerebral cavernous malformation protein critical for vascular integrity. J Biol Chem 2010;285:24099-107.
45. He Y, Zhang H, Yu L, et al. Stabilization of VEGFR2 signaling by cerebral cavernous malformation 3 is critical for vascular development. Sci Signal 2010;3:ra26.
46. Dibble CF, Horst JA, Malone MH, et al. Defining the functional domain of programmed cell death 10 through its interactions with phosphatidylinositol-3,4,5-trisphosphate. PLoS One 2010;5:e11740.
47. You C, Sandalcioglu IE, Dammann P, Felbor U, Sure U, Zhu Y. Loss of CCM3 impairs DLL4-Notch signalling: implication in endothelial angiogenesis and in inherited cerebral cavernous malformations. J Cell Mol Med 2013;17:407-18.
48. Wang Y, Liu H, Zhang Y, Ma D. cDNA cloning and expression of an apoptosis-related gene, humanTFAR15 gene. Sci China C Life Sci 1999;42:323-9.
49. Guclu B, Ozturk AK, Pricola KL, et al. Mutations in apoptosis-related gene, PDCD10, cause cerebral cavernous malformation 3. Neurosurgery 2005;57:1008-13.
50. Chen L, Tanriover G, Yano H, Friedlander R, Louvi A, Gunel M. Apoptotic functions of PDCD10/CCM3, the gene mutated in cerebral cavernous malformation 3. Stroke 2009;40:1474-81.
51. Akers AL, Johnson E, Steinberg GK, Zabramski JM, Marchuk DA. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): evidence for a two-hit mechanism of CCM pathogenesis. Hum Mol Genet 2009;18:919-30.
52. Gault J, Sain S, Hu LJ, Awad IA. Spectrum of genotype and clinical manifestations in cerebral cavernous malformations. Neurosurgery 2006;59:1278-84; discussion 1284.
53. Denier C, Labauge P, Brunereau L, et al. Sociéte Française de Neurochirgurgie. , Sociéte de Neurochirurgie de Langue Française. Clinical features of cerebral cavernous malformations patients with KRIT1 mutations. Ann Neurol 2004;55:213-20.
54. Denier C, Labauge P, Bergametti F, et al. Société Française de Neurochirurgie. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol 2006;60:550-6.
55. Riant F, Bergametti F, Fournier HD, et al. CCM3 Mutations are associated with early-onset cerebral hemorrhage and multiple meningiomas. Mol Syndromol 2013;4:165-72.
56. Shenkar R, Shi C, Rebeiz T, et al. Exceptional aggressiveness of cerebral cavernous malformation disease associated with PDCD10 mutations. Genet Med 2015;17:188-96.
57. Gianfrancesco F, Cannella M, Martino T, et al. Highly variable penetrance in subjects affected with cavernous cerebral angiomas (CCM) carrying novel CCM1 and CCM2 mutations. Am J Med Genet B Neuropsychiatr Genet 2007;144B:691-5.
58. Craig HD, Günel M, Cepeda O, et al. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum Mol Genet 1998;7:1851-8.
59. Vos IJ, Vreeburg M, Koek GH, van Steensel MA. Review of familial cerebral cavernous malformations and report of seven additional families. Am J Med Genet A 2017;173:338-51.
60. Battistini S, Rocchi R, Cerase A, et al. Clinical, magnetic resonance imaging, and genetic study of 5 Italian families with cerebral cavernous malformation. Arch Neurol 2007;64:843-8.
61. Scimone C, Donato L, Katsarou Z, Bostantjopoulou S, D'Angelo R, Sidoti A. Two novel KRIT1 and CCM2 mutations in patients affected by cerebral cavernous malformations: new information on CCM2 penetrance. Front Neurol 2018;9:953.
62. Gunel M, Awad IA, Finberg K, et al. A founder mutation as a cause of cerebral cavernous malformation in Hispanic Americans. N Engl J Med 1996;334:946-51.
63. Sahoo T, Johnson EW, Thomas JW, et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet 1999;8:2325-33.
64. Stahl S, Gaetzner S, Voss K, et al. Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Hum Mutat 2008;29:709-17.
65. Liquori CL, Penco S, Gault J, et al. Different spectra of genomic deletions within the CCM genes between Italian and American CCM patient cohorts. Neurogenetics 2008;9:25-31.
66. Cau M, Loi M, Melis M, et al. C329X in KRIT1 is a founder mutation among CCM patients in Sardinia. Eur J Med Genet 2009;52:344-8.
67. Gallione CJ, Solatycki A, Awad IA, Weber JL, Marchuk DA. A founder mutation in the Ashkenazi Jewish population affecting messenger RNA splicing of the CCM2 gene causes cerebral cavernous malformations. Genet Med 2011;13:662-6.
68. Ortiz L, Costa AF, Bellido ML, et al. Study of cerebral cavernous malformation in Spain and Portugal: high prevalence of a 14 bp deletion in exon 5 of MGC4607 (CCM2 gene). J Neurol 2007;254:322-6.
69. Mondéjar R, Solano F, Rubio R, et al. Mutation prevalence of cerebral cavernous malformation genes in Spanish patients. PLoS One 2014;9:e86286.
70. Perne A, Zhang X, Lehmann L, Groth M, Stuber F, Book M. Comparison of multiplex ligation-dependent probe amplification and real-time PCR accuracy for gene copy number quantification using the beta-defensin locus. Biotechniques 2009;47:1023-8.
71. Yao R, Yu T, Qing Y, Wang J, Shen Y. Evaluation of copy number variant detection from panel-based next-generation sequencing data. Mol Genet Genomic Med 2019;7:e00513.
72. Verlaan DJ, Siegel AM, Rouleau GA. Krit1 missense mutations lead to splicing errors in cerebral cavernous malformation. Am J Hum Genet 2002;70:1564-7.
73. Battistini S, Ricci C. Concern regarding classification of c.703G>A/p.Gly235Arg as a novel missense variant in KRIT1 gene. Hum Mutat 2020;41:1069-71.
74. Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol 1991;220:49-65.
75. Riant F, Odent S, Cecillon M, et al. Deep intronic KRIT1 mutation in a family with clinically silent multiple cerebral cavernous malformations. Clin Genet 2014;86:585-8.
76. Spiegler S, Rath M, Hoffjan S, et al. First large genomic inversion in familial cerebral cavernous malformation identified by whole genome sequencing. Neurogenetics 2018;19:55-9.
77. Labauge P, Denier C, Bergametti F, Tournier-lasserve E. Genetics of cavernous angiomas. Lancet Neurology 2007;6:237-44.
78. Spiegler S, Najm J, Liu J, et al. High mutation detection rates in cerebral cavernous malformation upon stringent inclusion criteria: one-third of probands are minors. Mol Genet Genomic Med 2014;2:176-85.
79. Rath M, Jenssen SE, Schwefel K, et al. High-throughput sequencing of the entire genomic regions of CCM1/KRIT1, CCM2 and CCM3/PDCD10 to search for pathogenic deep-intronic splice mutations in cerebral cavernous malformations. Eur J Med Genet 2017;60:479-84.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ricci C, Riolo G, Battistini S. Molecular genetic analysis of cerebral cavernous malformations: an update. Vessel Plus 2021;5:31. http://dx.doi.org/10.20517/2574-1209.2021.28
AMA Style
Ricci C, Riolo G, Battistini S. Molecular genetic analysis of cerebral cavernous malformations: an update. Vessel Plus. 2021; 5: 31. http://dx.doi.org/10.20517/2574-1209.2021.28
Chicago/Turabian Style
Ricci, Claudia, Giulia Riolo, Stefania Battistini. 2021. "Molecular genetic analysis of cerebral cavernous malformations: an update" Vessel Plus. 5: 31. http://dx.doi.org/10.20517/2574-1209.2021.28
ACS Style
Ricci, C.; Riolo G.; Battistini S. Molecular genetic analysis of cerebral cavernous malformations: an update. Vessel Plus. 2021, 5, 31. http://dx.doi.org/10.20517/2574-1209.2021.28
About This Article
Copyright

Data & Comments
Data


 Cite This Article 28 clicks
Cite This Article 28 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.