
Microvascular dysfunction following cardioplegic arrest and cardiopulmonary bypass


Abstract
Cardioplegia and cardiopulmonary bypass (CP/CPB) during cardiac surgery may cause systemic microvascular dysfunction. CP/CPB is associated with significant alterations in myogenic tone, agonist-induced vasomotor response, and endothelial function in various organs and vascular beds. These alterations can result in vessel spasm, organ malperfusion, and tissue damage. This review summarizes the current state of research in this field.
Keywords
INTRODUCTION
During cardiovascular surgery, cardioplegia (CP) and cardiopulmonary bypass (CPB) may influence in vivo vasomotor control and microvascular function, and as a result, affect organ perfusion. In general, basal perfusion increases immediately following surgery while the hyperemic response decreases. The diminished reactive hyperemic response has been hypothesized to be due to coronary vasodilatation that occurs due to tissue ischemia after surgery. In a study by Hiratzka et al.[1], the peak-to-resting flow velocity ratio following a coronary occlusion diminished from 4.4 ± 0.2 before bypass to 3.0 ± 0.3 after bypass. In a canine model reported in the same study, similar findings were apparent. During this same period, left ventricular perfusion increased, mean arterial pressure and coronary vascular resistance decreased, and myocardial oxygen consumption was unchanged.
These clinical and experimental studies suggest that major coronary vasodilatation occurring in the early period following CP/CPB when these modalities are used. In another study by De Backer et al.[2], hemodynamic and microcirculatory variables were examined before and after cardiac surgery with or without CPB. Vascular function was also examined in a non-cardiac surgical control group. Within hours of cardiac surgery, reductions in microvascular perfusion were more severe in patients undergoing CPB than in off-pump patients. Meanwhile, alterations in vascular function had normalized in the non-cardiac surgical controls at this time. In patients undergoing either on pump or off pump cardiac surgery, the microcirculatory alterations (i.e., extent of impaired microvascular perfusion and density of perfused vessels) decreased with time but persisted for more than 24 h after completion of the operation.
Interestingly, the degree of microvascular changes appeared to be related to tissue ischemia and correlated with the peak lactate levels after surgery. Myocardial blood flow and blood flow through other organs are critically important aspects of physiology to prevent ischemic injury. These are most accurately assessed with direct in vivo measurement. However, in vitro microvascular studies may allow us to better examine changes in specific vasomotor pathways and mechanisms compared to the in vivo assessment of organ perfusion. For example, when atrial and skeletal muscle arterioles harvested from patients were examined, the contractile response elicited by vasopressin in atrial coronary vessels was increased after surgery[3]. In contrast, the responses to norepinephrine, endothelin-1, and thromboxane A2 (TXA2) were decreased[4-7]. This demonstrates a differential effect of cardiovascular surgery on vascular reactivity depending on the agonist examined and even which vascular bed is examined. Thus, not only are in vitro vascular responses markedly abnormal after CP/CPB, but there are clinical in vivo correlates of these changes as well. Other studies also demonstrate potentially clinically important findings in the study of the microcirculation after cardiovascular surgery, namely disturbances in microcirculatory perfusion and adverse clinical outcomes[8,9].
This review will explore three different domains of microvascular dysfunction during and after CP/CPB, summarizing the current state of research and considering open possibilities for future investigations. The focus will be on post-CP/CPB alterations in myogenic tone, agonist-induced vasomotor tone, and endothelial dysfunction in the coronary and peripheral microvasculature.
MYOGENIC TONE AND CARDIOPLEGIA/CARDIOPULMONARY BYPASS
Background
Vascular tone is determined by many separate factors present in the various component of the blood vessel. These include the endothelium, vascular smooth muscle, surrounding tissue such as myocardium in the case of the heart, and neurohumoral factors. One factor affecting tissue perfusion is myogenic tone, critical for autoregulatory control of perfusion. Myogenic tone is an intrinsic property of vascular smooth muscle in many (but not all) vascular beds and plays an important role in regulation of the coronary and peripheral microcirculation (note that certain vascular beds, such as adipose arterioles, do not exhibit myogenic tone). Myogenic tone provides a baseline level of spontaneous vascular wall tone, allowing the microvasculature to respond to changes in blood flow and pressure. It also provides a baseline tonic level of vascular tone against which vasoactive mediators can modify vessel diameter. Changes in blood flow produce corresponding changes in microvascular resistance that are controlled by the contraction or relaxation of the arteriolar smooth muscle cells, with resulting changes in vessel diameter. Thus, myogenic tone often manifests as contraction of a vessel after a sustained increase in blood pressure triggers enough vasodilation to reach a critical point, after which vessel wall tension and transmural pressure respond with an elastic rebound effect. A key purpose of myogenic tone is likely to prevent unchecked passive dilation of vessels in response to increased intraluminal pressure[10].
Several mechanisms exist that regulate vascular myogenic tone. Ion channels such as TRPC6 and TRPM4 help mediate pressure-induced vascular smooth muscle depolarization[11]. Protein kinase C (PKC) serves as a regulator of the TRPM4 channel, and studies testing PKC inhibition have revealed attenuated pressure-induced myogenic vasoconstriction in human cerebral arteries[12]. PKC itself is sensitive to levels of intracellular calcium, with increases in intracellular calcium triggering heightened activation of PKC. Indeed, influx of calcium through voltage gated calcium channels has been shown to induce myogenic vasoconstriction in in vitro patch clamp experiments and intact vessel preparations[13,14]. Most myogenic responses are related to calcium influx that is thought to occur through L-type voltage gated calcium channels; however, T-type voltage gated calcium channels may also influence myogenic autoregulation in various vascular beds, as seen in animal models[15].
Epidermal growth factor receptor tyrosine kinase (EGFRtk) has been shown to affect coronary arteriole myogenic tone in coronary arterioles isolated from mouse models[16]. Intramural pressure leads to metalloproteinase 2/9 activation, release of heparin-binding EGF, and transactivation of EGFRtk[17]. Activation of EGFRtk by microvascular intraluminal pressure initiates a downstream intracellular signaling cascade involving JAK, STAT, Akt, and several other second messenger/effector molecules that ultimately results in enhanced myogenic tone/vasoconstriction. Likewise, the ERK1/2-JAK-STAT3 intracellular complexes have been implicated in myogenic tone of coronary arterioles[16].
Other studies implicate structural molecular components of vascular smooth muscle cells, including actin polymerization and cytoskeletal stabilization[18], and integrins[19] in the myogenic vascular responsiveness. Integrins specifically appear to be implicated in tyrosine phosphorylation cascades and mitogen activated protein kinase (MAPK) action, both of which are required for myogenic tone.
The effect of cardioplegia and cardiopulmonary bypass on myogenic tone
CP/CPB are associated with reduced coronary and peripheral microvascular myogenic tone across animal models and in patients[14,20-26]. These changes often manifest as post-CP/CPB systemic hypotension and in the extreme case may result in poor organ perfusion and injury. Several possible mechanisms have been put forward to explain this phenomenon. For example, blood cardioplegia and CPB reduces levels of ERK1/2 activation in coronary arterioles, which were correlated with reductions in myogenic tone before and after CP/CPB[22]. ERK1/2 regulates coronary vascular smooth muscle contraction, and pharmacologic inhibition of MEK1/2, a kinase upstream of ERK1/2, also results in diminished myogenic tone. PKC also influences ERK1/2 activation, suggesting that similar signal transduction cascades may be at play, affecting coronary myogenic tone[27,28].
Voltage-gated calcium channel function also appears altered in the microvasculature following CP/CPB. In human skeletal muscle arterioles, post-CPB myogenic tone has been associated with increased large conductance (BKCa) calcium-activated potassium channel activity[23]. Treatment with BKCa channel antagonist iberiotoxin significantly increased post-CPB microvascular myogenic tone; however, intermediate, and small conductance KCa channels appeared to have minimal effect on myogenic tone[23]. One result of protein kinase activation in vascular smooth muscle cells may be modulation of BKCa channel activity by phosphorylation of the channels themselves or assorted regulatory proteins[29]. Curiously, protein expression levels of BKCa channel were not altered in the microvascular smooth muscle following CPB, suggesting that posttranslational modifications to the channel (e.g., affecting open probability or duration) or alterations in downstream signaling are likely the driving force behind observed effects[23].
Either normothermic or hypothermic blood cardioplegia has been proposed as a potentially superior protocol to classic hypothermic crystalloid cardioplegia in preventing microvascular dysfunction and promoting myocardial protection during CP/CPB[10,30]. The rationale is that cold blood more closely approximates normal physiology and may provide more buffering capacity than cold crystalloid solution. However, debate continues concerning this topic, and little actual data conclusively demonstrates a clear clinical benefit of blood over crystalloid cardioplegia. In a porcine model of CPB comparing intermittent cold vs. continuous warm blood cardioplegia, both protocols of cardioplegia produced similar reductions in microvascular myogenic tone, suggesting that warm blood and intermittent cold cardioplegia have minimally different effects in that aspect[24]. However, other studies involving porcine models of hyperkalemic crystalloid cardioplegic solution vs. blood cardioplegia suggest that blood cardioplegia decreases myogenic tone to a lesser degree than cold crystalloid solution[25]. Clearly more studies, especially clinical studies involving patients, are needed before any definitive conclusions can be drawn.
Remaining questions
Several clinical variables that can potentially affect post-CPB changes in myogenic tone remain uninvestigated. For example, preoperative conditions such as diabetes may affect the outcomes of cardiovascular surgery. This is important because diabetes is a condition present in many patients undergoing coronary artery bypass grafting under CPB due to the high prevalence of coronary artery disease among this patient population[31]. While diabetes appears to alter myogenic tone in the microvasculature in humans post-CP/CPB, the specific mechanisms that drive these changes remain unclear[31]. Likewise, hypertension is another condition prevalent in many patients with cardiovascular disease. Hypertension may increase baseline myogenic tone and alter the structure and function of large arteries[32]; however, the specific contribution of preoperative hypertension to post-CPB myogenic tone remains to be elucidated. A similar lack of literature exists for the roles of age and gender.
AGONIST-INDUCED VASOMOTOR TONE AND CARDIOPLEGIA/CARDIOPULMONARY BYPASS
Agonist-induced vasomotor tone of the microvasculature refers to the contribution of neurotransmitters from the autonomic nervous system, and other vasoactive factors (e.g., hormones and neuropeptides), to the degree of tension within the vascular smooth muscle of arteries and arterioles. As with myogenic tone, CP/CPB has been shown to alter microvascular agonist-induced vasomotor tone, potentially contributing to observed postoperative microvascular dysfunction such as postoperative vasoplegia, hypotension, and organ malperfusion[10]. Dysfunctional agonist-induced vasomotor tone manifests differently in distinct vascular beds, presumably due to differences in receptor expression and other more distal mechanisms; specific details of any potential mechanisms remain to be elucidated. For example, consider decreased microvascular resistance in the peripheral and skeletal circulation versus increased propensity to spasm in the coronary, pulmonary, mesenteric, and cerebral circulations [Table 1][10,31].
Vasomotor agonist responsiveness following cardioplegic and cardiopulmonary bypass (CBP/CP)
| Agonist | Vasomodulatory effect | CPB/CP induced changes | Ref. |
| α-adrenergic agonist phenylephrine | Vasoconstriction | Diminished responsiveness in human coronary arterioles | [4,6] |
| β-adrenergic agonists | Vasodilation | Decreased responsiveness in human skeletal muscle arterioles | [36] |
| Neuropeptide Y | Coronary artery vasoconstriction and myocardial contraction | No changes in responsiveness; gender-based changes in human atrial tissue neuropeptide Y expression levels | [40,42] |
| Endothelin-1 | ETA receptor: vasoconstriction ETB2 receptor: nitric-oxide mediated vasodilation | Increased plasma levels of endothelin-1, immediate postoperative period Decreased responsiveness of human coronary and skeletal muscle arterioles | [5,43-47] |
| Serotonin | 5HT1B, 2A, and D receptors: vasoconstriction 5HT2B and 7 receptors: vasodilation | Impaired 5HT1B receptor mediated contractile response in skeletal vasculature Increased coronary and pulmonary vasoconstriction | [48-54] |
| Vasopressin | Vasoconstriction | Increased responsiveness in human coronary arterioles Decreased responsiveness in human skeletal muscle arterioles | [3] |
| Thromboxane A2 | Vasoconstriction | Reduced responsiveness in human coronary arterioles | [7,54] |
Autonomic nervous system: α- and β-adrenergic receptors, phenylephrine
The autonomic nervous system contribution to vasomotor tone relies in large part on the sympathetic nervous system, which controls vascular diameter through norepinephrine action and epinephrine on α- and β-adrenergic receptors located on vascular smooth muscle cells. α-adrenergic receptors are important vasomotor mediators of blood vessels across the body[33]. α-1 receptors operate through a G-protein coupled receptor (GPCR) mediated pathway involving phospholipase C, elevated intracellular calcium levels, and activation of PKC, all of which lead to smooth muscle contraction and vasoconstriction. α-2 receptors have more mixed effects and operate through a GPCR pathway that downregulates adenylyl cyclase, reducing levels of cyclic adenosine monophosphate (cAMP). Curiously, following CP/CPB, α adrenergic receptors do not show protein-level changes in expression[34]. However, experiments also show that post-CPB response to phenylephrine, an α-1 adrenergic agonist, is markedly diminished in human coronary arterioles[4]. Hence post-translational modification to receptors, alterations in receptor trafficking (resulting in fewer membrane bound receptors, even with an unchanged amount of total receptors) or changes in downstream signaling are likely mediating this altered α adrenergic response. This may be the result of receptor desensitization or impairment of the cellular pathway responsible for the contractile action due to activation of the α-1 adrenergic receptor.
β-adrenergic receptors are important vasodilators in the microvasculature of the myocardium, skeletal muscle and other tissues, acting through a GPCR mediated intracellular signal transduction cascade[35]. Elevated levels of cAMP result from β-2 receptor stimulation; this activates protein kinase A (PKA), which phosphorylates myosin light chain kinase thereby triggering smooth muscle relaxation. In vitro experiments on microvessels removed from human skeletal muscle show decreased responsiveness to β-2 receptor agonists following cardiopulmonary bypass, suggesting a postoperative deficit in β-2 receptor mediated vasodilation[36]. Given that β-2 receptor expression levels were unchanged, these changes are most likely mediated by changes to downstream elements of the β-2 receptor transduction cascade[36].
Neuropeptide Y
Neuropeptide Y is widely distributed in the central and peripheral nervous system, and often functions as a sympathetic co-stimulator alongside norepinephrine[37]. Neuropeptide Y is also abundant in the heart and coronary arteries, facilitating coronary artery constriction and myocardial contraction[38]. Of the many different neuropeptide Y receptors currently identified, the Y1, Y2, and Y5 receptors appear to be the most important regulators of cardiovascular homeostasis[38,39]. Each of these receptors function through a GPCR signal transduction cascade culminating with inhibition of adenylyl cyclase and reduced action of PKA. The Y1 receptor specifically may also act through a phospholipase C-dependent pathway to stimulate vasoconstriction via changes in intracellular calcium levels. Human skeletal muscle microvasculature has been shown to respond to neuropeptide Y agonists with dose-dependent manner vasoconstriction, raising the possibility that aberrations in neuropeptide Y could influence vasomotor dysfunction in the peripheral vasculature following CPB[40]. CP/CPB significantly altered the gene/protein expression of neuropeptide Y in human blood and myocardium[41]. Expression of neuropeptide Y in human atrial tissue following CPB appears to show a level of gender stratification: samples from men exhibited reduced post-CPB neuropeptide Y mRNA while samples from women exhibited increased neuropeptide Y mRNA[42]. However, there appears to be no change in the response of human skeletal microvessels to neuropeptide Y following CPB, along with no changes in Y1 receptor abundance in the microvessels[40]. Additional studies will be required to verify these results.
Endothelin 1
Endothelin-1 (ET-1) is an important vasomodulator secreted by endothelial cells[43,44]. ET-1 functions through two different types of GPCRs: ETA and ETB receptors. ETA and ETB2 receptors are found in vascular smooth muscle and trigger vasoconstriction, along with cell proliferation (ETA) and inflammation (ETA)[44]. Conversely, the ETB2 receptor is predominantly on endothelial cells and functions in nitric oxide mediated vasodilation[44]. Following CPB during coronary artery bypass grafting, plasma endothelin-1 levels in systemic arterial and pulmonary circulations increased between 50%-85% in the immediate postoperative period[45,46]. However, in vitro contractile response of human coronary and skeletal muscle arterioles to ET-1 was shown to be markedly decreased following cardiopulmonary bypass[5,47]. Molecular analysis revealed no significant changes in ET receptor expression levels, thereby implicating posttranslational modifications, dysregulated receptor trafficking, or downstream signaling effects as potential mechanisms of ET-1 driven vasomotor dysfunction via ETA receptors and PKC-α signaling pathways[5,47].
Serotonin
Serotonin, released in high levels during platelet activation, functions as an important vasomodulator in the circulation through action of various serotonin receptors (5HTRs). During acute coronary syndromes, serotonin released from platelets may cause further platelet aggregation and affect downstream vasomotor tone. Serotonin may promote vasoconstriction predominantly by means of the 5HT2A receptor, with contributions from the 5HT1B and D receptors[48]. Meanwhile, certain vessels such as human umbilical arteries and skeletal muscle arterioles have been shown to dilate in response to stimulation of other serotonin receptors, such as the 5HT2B and 5HT7 receptors[48]. In addition to vasomodulatory effects, serotonin is also capable of activating phospholipase A2 (PLA2), the key initial enzyme involved in arachidonic acid metabolism and generation of inflammatory mediators like prostaglandins, lipoxygenases, and thromboxane A2 (TXA2)[49,50]. Most of the serotonin receptors belong to the GPCR family.
CP/CPB impairs the 5HT1B receptor mediated contractile response to serotonin postoperatively in the skeletal vasculature[51]. However, there appear to be no changes in the expression levels of skeletal vascular 5HT1B receptors following CPB, indicating that the observed effects are likely due to posttranslational or downstream signaling modifications[51]. In contrast, serotonin-induced coronary and pulmonary vascular contraction have been shown to increase following cardioplegia-reperfusion and are associated with increased expression of the pro-inflammatory enzymes, PLA2 and cyclooxygenase 2 (COX2), key mediators of prostaglandin, thromboxane, and prostacyclin synthesis[52-55]. Furthermore, 5HT1B receptor mRNA expression increased following CPB in coronary arterial smooth muscle, suggesting that increased 5HT1B receptor expression may mediate a hypercontractile state in the coronary microcirculation following CPB.
Vasopressin
Vasopressin is released from the posterior pituitary gland during stress such as myocardial infarction and cardiogenic shock. In addition, vasopressin has long been used as a drug for maintaining systemic blood pressure and minimizing hypotension after cardiac surgery[56]. As the name suggests, vasopressin induces constriction of arterial vascular smooth muscle by binding to V1 receptors[57]. V1 receptors primarily function through GPCR pathways, inducing activation of PLA2, phospholipase C, and calcium influx, the latter being most relevant to vasoconstriction. Circulating vasopressin increase in a variety of pathologic states including hypotension, hemorrhagic shock, and hypoxia[3]. Experiments with human coronary arterioles revealed increased responses to vasopressin following CP/CPB, associated with increased postoperative expression of V1 receptors[3]. Curiously, human skeletal muscle arterioles exhibited decreased contractile response to vasopressin following CP/CPB[3]. Future studies are required to elucidate the specific mechanisms of altered vascular responsiveness to vasopressin in the coronary and peripheral microcirculations, although observed aberrations in PKC and MAPK systems (implicated in V1 receptor signal transduction), along with increased oxidative stress following CPB, may be at play[22,58,59].
Thromboxane A2
TXA2 is a pro-inflammatory, prothrombotic product of cyclooxygenase 1 and thromboxane synthase action on arachidonic acid and prostaglandin H-2[60]. With respect to vasomotor tone, TXA2 acts on thromboxane receptors of vascular smooth muscle, inducing activation of phospholipase C, release of intracellular calcium stores into the cytosol, and activation of myosin light chain kinase[60]. CPB induces a generalized systemic inflammatory state that contributes to vasomotor dysfunction, involving elevated levels of PLA2, TXA2, and thromboxane synthase, postoperatively[10,52,54]. High postoperative TXA2 may be responsible for heightened platelet activation and an overall prothrombotic state following CPB, a supposition reinforced by the use of thromboxane receptor antagonists and thromboxane synthase inhibitors to reduce post-CPB thrombosis[7]. However, despite higher TXA2 levels, the post-CPB contractile response of human coronary arterioles to TXA2 analogs is markedly impaired postoperatively[7]. It is possible that sustained high levels of TXA2 may induce decreased sensitivity to TXA2-mediated vascular smooth muscle constriction. Other elements of the post-CPB inflammatory response such as nitric oxide, free radicals, and activated neutrophils may also contribute to increased vascular permeability and diminished responsiveness to TXA2[7].
Further considerations and future directions
As with myogenic tone, several additional clinical variables can influence the effects of CP/CPB on agonist-induced vasomotor tone. Among patients with diabetes, post-CP/CPB attenuation of phenylephrine induced coronary arteriolar vasoconstriction, serotonin induced peripheral microvascular constriction, ET-1 induced peripheral vasoconstriction, and coronary nitric-oxide mediated vasodilation are more pronounced compared to the responses observed in non-diabetic patients[4,31,51]. Meanwhile, the coronary arteriolar contractile response to vasopressin exhibits more profound increases following CP/CPB in diabetic vs. nondiabetic patients[3]. Furthermore, studies are needed to examine the effect of other pervasive preexisting conditions, such as hypertension, hyperlipidemia, and clinical variables such as sex and age, on agonist-induced vasomotor tone following CP/CPB.
ENDOTHELIAL DYSFUNCTION AND CARDIOPLEGIA/CARDIOPULMONARY BYPASS
Endothelial dysfunction is a broad term referring to: (1) immune response induced alterations to the structure and permeability of the vascular endothelium; and (2) disturbances in the relative production/availability of nitric oxide, and other endothelium-derived relaxing and contracting factors[61]. During cardioplegia, administration of cardioplegic solution (often cold hyperkalemic solution) arrests myocardial contractility to facilitate open heart surgery, after which heart function is restored through reperfusion[62]. This process, and the accompanying ischemia-reperfusion, can damage the coronary endothelium that may have detrimental effects on postoperative cardiac function[59,63-69]. Moreover, endothelial injury extends far beyond the coronary circulation alone; the vascular beds of many organ systems, including the pulmonary and mesenteric circulations, sustain damage following CP/CPB[70-72].
Endothelial derived relaxing factor: nitric oxide
The vascular endothelium produces a vast array of vasoconstrictive and vasodilatory substances that influence vascular tone, immune cell/platelet-vessel wall interactions, and the overall health of vascular smooth muscle[73]. An important endothelium-derived vasodilatory substance is so-called endothelial-derived relaxing factor, which in arteries, veins and cultured vascular endothelial cells has been identified as nitric oxide. Nitric oxide functions as an important regulator of blood flow and tissue oxygenation by regulating vasomotor tone of vascular smooth muscle. In addition, nitric oxide inhibits mitochondrial reactive oxygen species production, inhibits inflammation, and quenches superoxide - thereby removing its own presence and creating peroxynitrite in its place. Normal, healthy endothelial cells exhibit constitutive activation of endothelial nitric oxide synthase (eNOS); activated endothelial cells, inflammatory cells, cardiomyocytes, and vascular smooth muscle cells express inducible nitric oxide synthase (iNOS)[10].
Both eNOS and iNOS produce nitric oxide as a byproduct of the conversion of L-arginine to L-citrulline. A gas, nitric oxide activates soluble guanylyl cyclase in the vascular smooth muscle, which in turn converts guanosine monophosphate to cyclic guanosine monophosphate, leading to activation of protein kinase G and phosphorylation of calcium channels[74]. This series of events results in vasodilation. Beyond vasomotor effects, nitric oxide also inhibits leukocyte adhesion and platelet activation[10]. Reduced activity of eNOS has been observed following CP/CPB, mediated by a variety of potential mechanisms including depletion of L-arginine, changes in intracellular calcium concentrations, and cell membrane injuries[59,75-79]. Generation of free radicals during reperfusion after cardioplegia leads to increased nitric oxide breakdown[80]. In addition, endothelial exposure to byproducts and mediators of inflammation, including activated complement and neutrophils, further attenuates the ability of vascular endothelium to generate nitric oxide following CP/CPB[81,82].
PGI2 and COX
Prostacyclin (PGI2) is an important vasodilator synthesized in vascular endothelium[50]. It also functions as a counterweight to the pro-thrombotic, vasoconstrictive effects of TXA2[50]. PGI2 is synthesized from prostaglandin H2 (PGH2) with the assistance of PGI synthase. PGH2 is itself a product of arachidonic acid metabolism, the result of cyclooxygenase (COX) enzyme action[50]. Two COX isoforms have been identified in human cells: COX1 and COX2. COX1 is a housekeeping isoform constitutively expressed in most cells of the body; COX2 expression is induced by inflammatory cytokines in immune cells and activated endothelial cells[83]. Studies have shown that enhanced activity of COX2 leads to a shift in favor of PGI2 over TXA2, while reduced COX2 activity (or predominant COX1 activity) produces the opposite effect[84-86]. Inflammatory cytokines and endothelial stress have been demonstrated to upregulate COX2 in in vitro models[50].
COX2 expression increases during reperfusion following CP/CPB in a variety of vascular beds, including coronary, cerebral, and pulmonary circulation. This observed effect is consistent with the principle of inflammation-induced changes in vascular endothelial cells[87,88]. Meanwhile, COX1 levels remain unchanged. Other experiments using skeletal muscle microvessels confirmed COX2 upregulation post-CPB vs. pre-CPB, and associate this with altered peripheral microvascular reactivity (e.g., enhanced bradykinin-induced relaxation responses)[89]. Despite enhanced PGI2 levels due to enhanced COX2 action, the overall effect of higher endothelial COX2 expression following CP/CPB appears to be more potent vasoconstriction, given how administration of COX2 inhibitors attenuates vasoconstriction and release of contractile prostanoids following CP/CPB[88].
Endothelium-derived hyperpolarizing factor
Endothelium-derived hyperpolarizing factor (EDHF) essentially refers to an endothelium-dependent pathway involving calcium, opening of specific potassium channels (e.g., the calcium-sensitive potassium channels such as KCa2.3 and KCa3.1), and vascular endothelial and smooth muscle hyperpolarization[90-93]. Such calcium-activated potassium channel (KCa) pathways are thought to play important roles in dilation of coronary arteries, and animal models have implicated bradykinin in activation of coronary artery KCa channels[66,94]. Even in peripheral arteries, intermediate and small conductance KCa channels have been demonstrated as mediators of vasodilation[95].
CP/CPB appears to induce changes in coronary and peripheral EDHF pathways/responsiveness. Studies on human atrial and coronary microvessels reveal significantly reduced post-reperfusion relaxation responses after activation of intermediate and small conductance KCa channels following CPB, despite no alterations in channel polypeptide or mRNA expression levels[96-98]. A similar situation of reduced responsiveness to KCa channel activation has been demonstrated in human skeletal muscle arterioles following CPB[95]. Pharmacologic intervention in the EDHF pathway has shown promise for mitigating endothelial dysfunction following CP/CPB. For example, in animal and human coronary artery endothelial cells, administration of a small conductance calcium-activated potassium channel activator before and during cardioplegic hypoxia significantly improved overall coronary endothelial function, assessed by preserved responsiveness to bradykinin, ADP, and substance P[98-100].
Immune activation and oxidative stress
Activation of the complement system and coagulation cascades occur when blood encounters the CPB circuit[101,102]. Complement factors (e.g., anaphylaxins C3a and C5a) can induce synthesis of pro-inflammatory cytokines, like IL-1, IL-6, and TNF-α which in turn stimulate endothelial changes that facilitate neutrophil chemotaxis, extravasation, and tissue edema. Activation of the classical complement pathway specifically may cause direct vascular endothelial injury[103]. The terminal membrane attack complex formed from complement fragments C5b-9 has also been shown to cause direct endothelial injury in porcine coronary and mesenteric arteries following ischemia-reperfusion and CPB[82,104]. Administration of anti-C5a antibodies before onset of CPB in porcine model exhibits reduced coronary/pulmonary endothelial dysfunction assessed through degree of impairment of endothelial relaxation to various agonists (e.g., clonidine)[105,106].
Leukocyte activation may also damage vascular endothelium during and after CPB through production of reactive oxygen species, proteolytic enzymes, and inflammatory cytokines[10,107]. Increased expression of endothelial selectins after onset of CPB allows for increased neutrophil tissue infiltration across vascular walls. Further evidence for a role for leukocytes in post-CPB endothelial dysfunction comes from use of leukocyte-depleted (or inhibited) blood for reperfusion following cardioplegia in pig models, which demonstrated improved postoperative myocardial perfusion, along with improved coronary and cerebral vascular function[107-109]. Finally, in porcine model of CPB, addition of superoxide dismutase significantly preserved the vasomotor responses of coronary microvessels to serotonin and ADP, suggesting a role for oxygen derived free radicals in endothelial dysfunction[59,63].
Endothelial barrier integrity
CPB reduces the integrity of endothelial barrier function by compromising endothelial adherens junctions, leading to increased vascular permeability[31,110]. Animal and human studies demonstrate that CP/CPB is associated with increases in VEGF and/or its receptors in blood and tissues and enhanced tissue edema[111-113]. In porcine myocardial tissue following CPB, levels of small molecular weight fragments of VE cadherin, β-catenin, and gamma-catenin increased relative to pre-CPB levels, indicating enhanced protein degradation[114,115].
Experiments in human skeletal muscle arterioles and right atrial tissue report increased phosphorylation of vascular endothelial cadherins, along with overall decreases β-catenin levels[116,117]. Thus, one possible mechanism of disrupted endothelial barrier integrity might be enhanced calcium-calmodulin dependent activation of myosin light chain kinases and Rho GTPases, which in turn phosphorylate cadherins and catenins, tagging them for degradation[118]. The angiopoietin-Tie2 ligand receptor system may also be implicated in endothelial barrier dysfunction following CPB. Angiopoietin 2 binding to Tie2 receptor increases the responsiveness of the vascular endothelium to pro-inflammatory cytokines, and levels of angiopoietin 2 have been shown to increase in serum following CPB[119].
Future considerations and directions
Among patients undergoing CP/CPB and cardiac surgery, further work needs to be done to examine the effects of other comorbidities on postoperative endothelial dysfunction. A good amount of work has been done in patients and animals with diabetes, with studies suggesting that declines in overall coronary and peripheral microvascular reactivity following CP/CPB are more pronounced in diabetic versus nondiabetic patients[31,116,120-122]. More specific differences between diabetic and nondiabetic patients after CP/CPB include enhanced upregulation of COX2 and enhanced inactivation of small and intermediate calcium-sensitive potassium channels[89,97,123]. Next, even though hypertension has been shown to cause significant changes to the microvasculature through mechanisms such as heightened reactive oxygen species production and perivascular inflammation, little work has been done to investigate specific contributions of pre-existing hypertension in vasomotor/endothelial dysfunction following CPB[124,125]. Similarly, the contributions of other preexisting conditions, such as hyperlipidemia, and other clinical variables such as age and sex deserve further investigation. Most previous studies on microvascular function were conducted in the immediate aftermath of CP/CPB and cardiac surgery. However, it is largely unknown how long the CP/CPB-driven endothelial and vasomotor changes persist. We hypothesize that many of these changes will last for several weeks post-CP/CPB; however, further studies are necessary to confirm any hypotheses.
CONCLUSION
Vascular changes that occur in most vascular beds during surgery that may lead to altered myogenic, agonist-induced and neurohumoral control of organ perfusion [Figure 1]. These microvascular and macrovascular changes occur due to inflammatory influence such as increased leukocytes and cytokines, increased oxidative stress, complement activation, and other factors such as receptor denaturation. Endothelium, vascular smooth muscle and extravascular cells may all be affected. These changes may affect organ perfusion and lead to myocardial infarction, stroke and renal failure. A better understanding of the effects of cardiac surgery on the vasomotor regulation and other forms of microvascular dysfunction after surgery may allow us to better care for patients after cardiac surgery and improve outcomes.
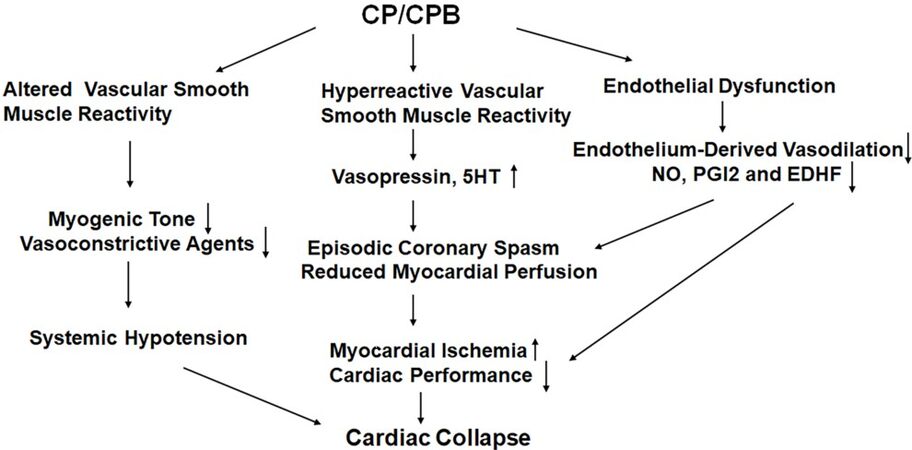
Figure 1. Schematic of general alterations in microvascular reactivity after cardioplegia (CP) and cardiopulmonary bypass (CPB). 5-HT: Serotonin; NO: nitric oxide; PGI2: prostaglandin I2; EDHF: endothelium-derived hyperpolarizing factor. Side arrows indicate overall direction of specific changes.
DECLARATIONS
Authors’ contributionsMade substantial contributions to the conception and design of this review article: Feng J, Kant S, Sellke FW
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis research project was supported by the National Institute of Health (NIH): R01-HL46716 to Sellke FW, 1R01HL127072-01A1 to Feng J, and 1R01 HL136347-01 to Feng J.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Hiratzka LF, Eastham CL, Carter JG, et al. The effects of cardiopulmonary bypass and cold cardioplegia on coronary flow velocity and the reactive hyperemic response in patients and dogs. Ann Thorac Surg 1988;45:474-81.
2. De Backer D, Dubois MJ, Schmartz D, et al. Microcirculatory alterations in cardiac surgery: effects of cardiopulmonary bypass and anesthesia. Ann Thorac Surg 2009;88:1396-403.
3. Sellke N, Kuczmarski A, Lawandy I, et al. Enhanced coronary arteriolar contraction to vasopressin in patients with diabetes after cardiac surgery. J Thorac Cardiovasc Surg 2018;156:2098-107.
4. Sellke N, Gordon C, Lawandy I, et al. Impaired coronary contraction to phenylephrine after cardioplegic arrest in diabetic patients. J Surg Res 2018;230:80-6.
5. Feng J, Chu LM, Robich MP, et al. Effects of cardiopulmonary bypass on endothelin-1-induced contraction and signaling in human skeletal muscle microcirculation. Circulation 2010;122:S150-5.
6. Sodha NR, Feng J, Clements RT, et al. Protein kinase C alpha modulates microvascular reactivity in the human coronary and skeletal microcirculation. Surgery 2007;142:243-52.
7. Feng J, Liu Y, Chu LM, et al. Thromboxane-induced contractile response of human coronary arterioles is diminished after cardioplegic arrest. Ann Thorac Surg 2011;92:829-36.
8. Murkin JM. Monitoring and optimization of the microcirculation during CPB. J Thorac Dis 2019;11:S1489-91.
9. Os MM, van den Brom CE, van Leeuwen ALI, Dekker NAM. Microcirculatory perfusion disturbances following cardiopulmonary bypass: a systematic review. Crit Care 2020;24:218.
10. Ruel M, Khan TA, Voisine P, Bianchi C, Sellke FW. Vasomotor dysfunction after cardiac surgery. Eur J Cardiothorac Surg 2004;26:1002-14.
11. Jackson WF. Ion channels and the regulation of myogenic tone in peripheral arterioles. Ion Channels and Calcium Signaling in the Microcirculation. Curr Top Membr 2020;85:19-58.
12. Earley S, Straub SV, Brayden JE. Protein kinase C regulates vascular myogenic tone through activation of TRPM4. Am J Physiol Heart Circ Physiol 2007;292:H2613-22.
13. Schubert R, Brayden JE. . Stretch-activated Cation Channels and the Myogenic Response of Small Arteries. In: Kamkin A, Kiseleva I, editors. Mechanosensitivity in Cells and Tissues. Moscow: Academia; 2005.
14. Dessy C, Matsuda N, Hulvershorn J, Sougnez CL, Sellke FW, Morgan KG. Evidence for involvement of the PKC-alpha isoform in myogenic contractions of the coronary microcirculation. Am J Physiol Heart Circ Physiol 2000;279:H916-23.
15. Jensen LJ, Nielsen MS, Salomonsson M, Sørensen CM. T-type Ca2+ channels and autoregulation of local blood flow. Channels (Austin) 2017;11:183-95.
16. Amin AH, Abd Elmageed ZY, Partyka M, Matrougui K. Mechanisms of myogenic tone of coronary arteriole: Role of down stream signaling of the EGFR tyrosine kinase. Microvasc Res 2011;81:135-42.
17. Lucchesi PA, Sabri A, Belmadani S, Matrougui K. Involvement of metalloproteinases 2/9 in epidermal growth factor receptor transactivation in pressure-induced myogenic tone in mouse mesenteric resistance arteries. Circulation 2004;110:3587-93.
18. Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J 2002;16:72-6.
19. Davis MJ, Wu X, Nurkiewicz TR, et al. Integrins and mechanotransduction of the vascular myogenic response. Am J Physiol Heart Circ Physiol 2001;280:H1427-33.
20. Wang SY, Friedman M, Franklin A, Sellke FW. Myogenic reactivity of coronary resistance arteries after cardiopulmonary bypass and hyperkalemic cardioplegia. Circulation 1995;92:1590-6.
21. Wang SY, Stamler A, Li J, Johnson RG, Sellke FW. Decreased myogenic reactivity in skeletal muscle arterioles after hypothermic cardiopulmonary bypass. J Surg Res 1997;69:40-4.
22. Khan TA, Bianchi C, Ruel M, et al. Mitogen-activated protein kinase inhibition and cardioplegia-cardiopulmonary bypass reduce coronary myogenic tone. Circulation 2003;108 Suppl 1:II348-53.
23. Feng J, Liu Y, Khabbaz KR, et al. Large conductance calcium-activated potassium channels contribute to the reduced myogenic tone of peripheral microvasculature after cardiopulmonary bypass. J Surg Res 2009;157:123-8.
24. Tofukuji M, Stamler A, Li J, Hariawala MD, Franklin A, Sellke FW. Comparative effects of continuous warm blood and intermittent cold blood cardioplegia on coronary reactivity. Ann Thorac Surg 1997;64:1360-7.
25. Wang SY, Stamler A, Tofukuji M, Deuson TE, Sellke FW. Effects of blood and crystalloid cardioplegia on adrenergic and myogenic vascular mechanisms. Ann Thorac Surg 1997;63:41-9.
26. Banitt PF, Dai HB, Wang SY, Friedman M, Sellke FW. Myogenic and agonist induced responses of coronary venules after cold hyperkalaemic cardioplegia. Cardiovasc Res 1995;29:827-33.
27. Hattori Y, Kakishita H, Akimoto K, Matsumura M, Kasai K. Glycated serum albumin-induced vascular smooth muscle cell proliferation through activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by protein kinase C. Biochem Biophys Res Commun 2001;281:891-6.
28. Velarde V, Jenkins AJ, Christopher J, Lyons TJ, Jaffa AA. Activation of MAPK by modified low-density lipoproteins in vascular smooth muscle cells. J Appl Physiol (1985) 2001;91:1412-20.
29. Liu Y, Terata K, Chai Q, Li H, Kleinman LH, Gutterman DD. Peroxynitrite inhibits Ca2+-activated K+ channel activity in smooth muscle of human coronary arterioles. Circ Res 2002;91:1070-6.
30. Nardi P, Pisano C, Bertoldo F, et al. Warm blood cardioplegia versus cold crystalloid cardioplegia for myocardial protection during coronary artery bypass grafting surgery. Cell Death Discov 2018;4:23.
31. Feng J, Sellke F. Microvascular dysfunction in patients with diabetes after cardioplegic arrest and cardiopulmonary bypass. Curr Opin Cardiol 2016;31:618-24.
32. Pires PW, Jackson WF, Dorrance AM. Regulation of myogenic tone and structure of parenchymal arterioles by hypertension and the mineralocorticoid receptor. Am J Physiol Heart Circ Physiol 2015;309:H127-36.
33. Brummelen P, Jie K, van Zwieten PA. Alpha-adrenergic receptors in human blood vessels. Br J Clin Pharmacol 1986;21 Suppl 1:33S-9S.
34. Wang SY, Friedman M, Johnson RG, Weintraub RM, Sellke FW. Adrenergic regulation of coronary microcirculation after extracorporeal circulation and crystalloid cardioplegia. Am J Physiol 1994;267:H2462-70.
35. Lundvall J, Järhult J. Beta adrenergic dilator component of the sympathetic vascular response in skeletal muscle. Influence on the micro-circulation and on transcapillary exchange. Acta Physiol Scand 1976;96:180-92.
36. Ziegler O, Anderson K, Liu Y, et al. Skeletal muscle microvasculature response to β-adrenergic stimuli is diminished with cardiac surgery. Surgery 2020;167:493-8.
37. Tan CMJ, Green P, Tapoulal N, Lewandowski AJ, Leeson P, Herring N. The role of neuropeptide Y in cardiovascular health and disease. Front Physiol 2018;9:1281.
39. Silva AP, Cavadas C, Grouzmann E. Neuropeptide Y and its receptors as potential therapeutic drug targets. Clinica Chimica Acta 2002;326:3-25.
40. Mirman B, Ikeda I, Zhang Z, et al. Effects of neuropeptide Y on the microvasculature of human skeletal muscle. Surgery 2020;168:155-9.
41. Matyal R, Mahmood F, Robich M, et al. Chronic type II diabetes mellitus leads to changes in neuropeptide Y receptor expression and distribution in human myocardial tissue. Eur J Pharmacol 2011;665:19-28.
42. Meng F, Han J, Wang J, Zhang H, Xu C, Meng X. The gender-specific expression of neuropeptide Y and neuropeptide Y receptors in human atrial tissue during cardiopulmonary bypass surgery. J Thorac Dis 2018;10:6563-8.
43. Marasciulo FL, Montagnani M, Potenza MA. Endothelin-1: the yin and yang on vascular function. Curr Med Chem 2006;13:1655-65.
44. Kowalczyk A, Kleniewska P, Kolodziejczyk M, Skibska B, Goraca A. The role of endothelin-1 and endothelin receptor antagonists in inflammatory response and sepsis. Arch Immunol Ther Exp (Warsz) 2015;63:41-52.
45. Bond BR, Dorman BH, Clair MJ, et al. Endothelin-1 during and after cardiopulmonary bypass: association to graft sensitivity and postoperative recovery. J Thorac Cardiovasc Surg 2001;122:358-64.
46. Verma S, Maitland A, Weisel RD, et al. Increased endothelin-1 production in diabetic patients after cardioplegic arrest and reperfusion impairs coronary vascular reactivity: reversal by means of endothelin antagonism. J Thorac Cardiovasc Surg 2002;123:1114-9.
47. Feng J, Liu Y, Khabbaz KR, et al. Endothelin-1-induced contractile responses of human coronary arterioles via endothelin-A receptors and PKC-alpha signaling pathways. Surgery 2010;147:798-804.
48. Watts SW, Morrison SF, Davis RP, Barman SM. Serotonin and blood pressure regulation. Pharmacol Rev 2012;64:359-88.
49. Berg KA, Clarke WP. Regulation of 5-HT1A and 5-HT1B receptor systems by phospholipid signaling cascades. Brain Res Bull 2001;56:471-7.
50. Caughey GE, Cleland LG, Penglis PS, Gamble JR, James MJ. Roles of cyclooxygenase (COX)-1 and COX-2 in prostanoid production by human endothelial cells: selective up-regulation of prostacyclin synthesis by COX-2. J Immunol 2001;167:2831-8.
51. Sabe SA, Feng J, Liu Y, Scrimgeour LA, Ehsan A, Sellke FW. Decreased contractile response of peripheral arterioles to serotonin after CPB in patients with diabetes. Surgery 2018;164:288-93.
52. Metais C, Li J, Simons M, Sellke FW. Serotonin-induced coronary contraction increases after blood cardioplegia-reperfusion: role of COX-2 expression. Circulation 1999;100:II328-34.
53. Metais C, Bianchi C, Li J, Simons M, Sellke FW. Serotonin-induced human coronary microvascular contraction during acute myocardial ischemia is blocked by COX-2 inhibition. Basic Res Cardiol 2001;96:59-67.
54. Robich MP, Araujo EG, Feng J, et al. Altered coronary microvascular serotonin receptor expression after coronary artery bypass grafting with cardiopulmonary bypass. J Thorac Cardiovasc Surg 2010;139:1033-40.
55. Sato K, Li J, Metais C, Bianchi C, Sellke F. Increased pulmonary vascular contraction to serotonin after cardiopulmonary bypass: role of cyclooxygenase. J Surg Res 2000;90:138-43.
56. Masetti P, Murphy SF, Kouchoukos NT. Vasopressin therapy for vasoplegic syndrome following cardiopulmonary bypass. J Card Surg 2002;17:485-9.
57. Holmes CL, Landry DW, Granton JT. Science review: Vasopressin and the cardiovascular system part 1--receptor physiology. Crit Care 2003;7:427-34.
58. Khan TA, Bianchi C, Ruel M, Feng J, Sellke FW. Differential effects on the mesenteric microcirculatory response to vasopressin and phenylephrine after cardiopulmonary bypass. J Thorac Cardiovasc Surg 2007;133:682-8.
59. Sellke FW, Friedman M, Dai HB, Shafique T, Schoen FJ, Weintraub RM, Johnson RG. Mechanisms causing coronary microvascular dysfunction following crystalloid cardioplegia and reperfusion. Cardiovasc Res 1993;27:1925-32.
60. Nakahata N. Thromboxane A2: physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol Ther 2008;118:18-35.
61. Hadi HA, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc Health Risk Manag 2005;1:183-98.
62. He GW. Coronary endothelial function in open heart surgery. Clin Exp Pharmacol Physiol 1997;24:955-7.
63. Sellke FW, Shafique T, Ely DL, Weintraub RM. Coronary endothelial injury after cardiopulmonary bypass and ischemic cardioplegia is mediated by oxygen-derived free radicals. Circulation 1993;88:II395-400.
64. Sellke FW, Shafique T, Schoen FJ, Weintraub RM. Impaired endothelium-dependent coronary microvascular relaxation after cold potassium cardioplegia and reperfusion. J Thorac Cardiovasc Surg 1993;105:52-8.
65. Sellke FW, Shafique T, Johnson RG, Dai HB, Banitt PF, Schoen FJ, Weintraub RM. Blood and albumin cardioplegia preserve endothelium-dependent microvascular responses. Ann Thorac Surg 1993;55:977-85.
66. Feng J, Sellke ME, Ramlawi B, et al. Bradykinin induces microvascular preconditioning through the opening of calcium-activated potassium channels. Surgery 2006;140:192-7.
67. Feng J, Bianchi C, Li J, Sellke FW. Improved profile of bad phosphorylation and caspase 3 activation after blood versus crystalloid cardioplegia. Ann Thorac Surg 2004;77:1384-9; discussion 1389.
68. Feng J, Bianchi C, Sandmeyer JL, Sellke FW. Bradykinin preconditioning improves the profile of cell survival proteins and limits apoptosis after cardioplegic arrest. Circulation 2005;112:I190-5.
69. Feng J, Bianchi C, Li J, Sellke FW. Bradykinin preconditioning preserves coronary microvascular reactivity during cardioplegia-reperfusion. Ann Thorac Surg 2005;79:911-6.
70. Paparella D, Yau TM, Young E. Cardiopulmonary bypass induced inflammation: pathophysiology and treatment. An update. Eur J Cardiothorac Surg 2002;21:232-44.
71. Aljure OD, Fabbro M 2nd. Cardiopulmonary bypass and inflammation: the hidden enemy. J Cardiothorac Vasc Anesth 2019;33:346-7.
72. Levy JH, Tanaka KA. Inflammatory response to cardiopulmonary bypass. Ann Thorac Surg 2003;75:S715-20.
73. Lambert J, Aarsen M, Donker AJ, Stehouwer CD. Endothelium-dependent and -independent vasodilation of large arteries in normoalbuminuric insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol 1996;16:705-11.
74. Chen K, Pittman RN, Popel AS. Nitric oxide in the vasculature: where does it come from and where does it go? Antioxid Redox Signal 2008;10:1185-98.
75. Engelman DT, Watanabe M, Engelman RM, et al. Constitutive nitric oxide release is impaired after ischemia and reperfusion. J Thorac Cardiovasc Surg 1995;110:1047-53.
76. He G, Yang C. Hyperkalemia alters endothelium-dependent relaxation through non-nitric oxide and noncyclooxygenase pathway: a mechanism for coronary dysfunction due to cardioplegia. Ann Thorac Surg 1996;61:1394-9.
77. Andrási TB, Soós P, Bakos G, et al. L-arginine protects the mesenteric vascular circulation against cardiopulmonary bypass-induced vascular dysfunction. Surgery 2003;134:72-9.
78. Meldrum DR, Cleveland JC, Sheridan BC, Rowland RT, Banerjee A, Harken AH. Cardiac surgical implications of calcium dyshomeostasis in the heart. Ann Thorac Surg 1996;61:1273-80.
79. Mehlhorn U, Bloch W. Role of cardiopulmonary bypass and cardioplegic arrest in the regulation of cardiac nitric oxide synthase activity. J Thorac Cardiovasc Surg 2002;124:418-9.
80. Kukreja RC, Hess ML. The oxygen free radical system: from equations through membrane-protein interactions to cardiovascular injury and protection. Cardiovasc Res 1992;26:641-55.
81. Sellke FW, Boyle EM, Verrier ED. Endothelial cell injury in cardiovascular surgery: The pathophysiology of vasomotor dysfunction. Ann Thorac Surg 1996;62:1222-8.
82. Stahl GL, Reenstra WR, Frendl G. Complement-mediated loss of endothelium-dependent relaxation of porcine coronary arteries. Role of the terminal membrane attack complex. Circ Res 1995;76:575-83.
83. Crofford LJ. COX-1 and COX-2 tissue expression: implications and predictions. J Rheumatol Suppl 1997;49:15-9.
84. Matsumoto H, Naraba H, Murakami M, et al. Concordant induction of prostaglandin E2 synthase with cyclooxygenase-2 leads to preferred production of prostaglandin E2 over thromboxane and prostaglandin D2 in lipopolysaccharide-stimulated rat peritoneal macrophages. Biochem Biophys Res Commun 1997;230:110-4.
85. Brock TG, McNish RW, Peters-Golden M. Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J Biol Chem 1999;274:11660-6.
86. Ricciotti E, Yu Y, Grosser T, Fitzgerald GA. COX-2, the dominant source of prostacyclin. Proc Natl Acad Sci U S A 2013;110:E183.
87. Sodha NR, Clements RT, Sellke FW. Vascular changes after cardiac surgery: role of NOS, COX, kinases, and growth factors. Front Biosci (Landmark Ed) 2009;14:689-98.
88. Sellke FW. Vascular changes after cardiopulmonary bypass and ischemic cardiac arrest: roles of nitric oxide synthase and cyclooxygenase. Braz J Med Biol Res 1999;32:1345-52.
89. Feng J, Anderson K, Singh AK, et al. Diabetes upregulation of cyclooxygenase 2 contributes to altered coronary reactivity after cardiac surgery. Ann Thorac Surg 2017;104:568-76.
90. Garland CJ, Yarova PL, Jiménez-Altayó F, Dora KA. Vascular hyperpolarization to β-adrenoceptor agonists evokes spreading dilatation in rat isolated mesenteric arteries. Br J Pharmacol 2011;164:913-21.
91. Liu Y, Xie A, Singh AK, et al. Inactivation of endothelial small/intermediate conductance of calcium-activated potassium channels contributes to coronary arteriolar dysfunction in diabetic patients. J Am Heart Assoc 2015;4:e002062.
92. Liu Y, Kabakov AY, Xie A, et al. Metabolic regulation of endothelial SK channels and human coronary microvascular function. Int J Cardiol 2020;312:1-9.
93. Xing H, Zhang Z, Shi G, et al. Chronic inhibition of mROS protects against coronary endothelial dysfunction in mice with diabetes. Front Cell Dev Biol 2021;9:643810.
94. Si H, Heyken WT, Wölfle SE, et al. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ Res 2006;99:537-44.
95. Liu Y, Sellke EW, Feng J, et al. Calcium-activated potassium channels contribute to human skeletal muscle microvascular endothelial dysfunction related to cardiopulmonary bypass. Surgery 2008;144:239-44.
96. Feng J, Liu Y, Clements RT, et al. Calcium-activated potassium channels contribute to human coronary microvascular dysfunction after cardioplegic arrest. Circulation 2008;118:S46-51.
97. Liu Y, Cole V, Lawandy I, Ehsan A, Sellke FW, Feng J. Decreased coronary arteriolar response to KCa channel opener after cardioplegic arrest in diabetic patients. Mol Cell Biochem 2018;445:187-94.
98. Zhang Z, Shi G, Liu Y, et al. Coronary endothelial dysfunction prevented by small-conductance calcium-activated potassium channel activator in mice and patients with diabetes. J Thorac Cardiovasc Surg 2020;160:e263-80.
99. Yang Q, Huang JH, Man YB, Yao XQ, He GW. Use of intermediate/small conductance calcium-activated potassium-channel activator for endothelial protection. J Thorac Cardiovasc Surg 2011;141:501-10, 510.e1.
100. Yang Q, Underwood MJ, He GW. Calcium-activated potassium channels in vasculature in response to ischemia-reperfusion. J Cardiovasc Pharmacol 2012;59:109-15.
101. Chenoweth DE, Cooper SW, Hugli TE, Stewart RW, Blackstone EH, Kirklin JW. Complement activation during cardiopulmonary bypass: evidence for generation of C3a and C5a anaphylatoxins. N Engl J Med 1981;304:497-503.
102. Bruins P, te Velthuis H, Yazdanbakhsh AP, et al. Activation of the complement system during and after cardiopulmonary bypass surgery: postsurgery activation involves C-reactive protein and is associated with postoperative arrhythmia. Circulation 1997;96:3542-8.
103. Pinckard RN, Olson MS, Giclas PC, Terry R, Boyer JT, O'Rourke RA. Consumption of classical complement components by heart subcellular membranes in vitro and in patients after acute myocardial infarction. J Clin Invest 1975;56:740-50.
104. Tofukuji M, Stahl GL, Metais C, et al. Mesenteric dysfunction after cardiopulmonary bypass: role of complement C5a. Ann Thorac Surg 2000;69:799-807.
105. Park KW, Tofukuji M, Metais C, et al. Attenuation of endothelium-dependent dilation of pig pulmonary arterioles after cardiopulmonary bypass is prevented by monoclonal antibody to complement C5a. Anesth Analg 1999;89:42-8.
106. Tofukuji M, Stahl GL, Agah A, Metais C, Simons M, Sellke FW. Anti-C5a monoclonal antibody reduces cardiopulmonary bypass and cardioplegia-induced coronary endothelial dysfunction. J Thorac Cardiovasc Surg 1998;116:1060-8.
107. Tofukuji M, Metais C, Collard CD, et al. Effect of sialyl Lewisx oligosaccharide on myocardial and cerebral injury in the pig. Ann Thorac Surg 1999;67:112-9.
108. Kawata H, Sawatari K, Mayer JE, Jr. . Evidence for the role of neutrophils in reperfusion injury after cold cardioplegic ischemia in neonatal lambs. J Thorac Cardiovasc Surg. 1992;103:908-17; discussion 17-8.
109. Schermerhorn ML, Tofukuji M, Khoury PR, et al. Sialyl lewis oligosaccharide preserves cardiopulmonary and endothelial function after hypothermic circulatory arrest in lambs. J Thorac Cardiovasc Surg 2000;120:230-7.
110. Giacinto O, Satriano U, Nenna A, et al. Inflammatory response and endothelial dysfunction following cardiopulmonary bypass: pathophysiology and pharmacological targets. Recent Pat Inflamm Allergy Drug Discov 2019;13:158-73.
111. Tofukuji M, Metais C, Li J, Franklin A, Simons M, Sellke FW. Myocardial VEGF expression after cardiopulmonary bypass and cardioplegia. Circulation 1998;98:II242-6; discussion II7.
112. Emani S, Ramlawi B, Sodha NR, Li J, Bianchi C, Sellke FW. Increased vascular permeability after cardiopulmonary bypass in patients with diabetes is associated with increased expression of vascular endothelial growth factor and hepatocyte growth factor. J Thorac Cardiovasc Surg 2009;138:185-91.
113. Mieno S, Ramlawi B, Boodhwani M, et al. Role of stromal-derived factor-1alpha in the induction of circulating CD34+CXCR4+ progenitor cells after cardiac surgery. Circulation 2006;114:I186-92.
114. Bianchi C, Araujo EG, Sato K, Sellke FW. Biochemical and structural evidence for pig myocardium adherens junction disruption by cardiopulmonary bypass. Circulation 2001;104:I319-24.
115. Khan TA, Bianchi C, Araujo E, et al. Aprotinin preserves cellular junctions and reduces myocardial edema after regional ischemia and cardioplegic arrest. Circulation 2005;112:I196-201.
116. Feng J, Liu Y, Sabe AA, et al. Differential impairment of adherens-junction expression/phosphorylation after cardioplegia in diabetic versus non-diabetic patients. Eur J Cardiothorac Surg 2016;49:937-43.
117. Feng J, Liu Y, Singh AK, et al. Effects of diabetes and cardiopulmonary bypass on expression of adherens junction proteins in human peripheral tissue. Surgery 2017;161:823-9.
118. Hinsbergh VW, van Nieuw Amerongen GP. Endothelial hyperpermeability in vascular leakage. Vascul Pharmacol 2002;39:171-2.
119. Clajus C, Lukasz A, David S, et al. Angiopoietin-2 is a potential mediator of endothelial barrier dysfunction following cardiopulmonary bypass. Cytokine 2012;60:352-9.
120. Feng J, Liu Y, Chu LM, et al. Changes in microvascular reactivity after cardiopulmonary bypass in patients with poorly controlled versus controlled diabetes. Circulation 2012;126:S73-80.
121. Feng J, Chu LM, Dobrilovic N, Liu Y, Singh AK, Sellke FW. Decreased coronary microvascular reactivity after cardioplegic arrest in patients with uncontrolled diabetes mellitus. Surgery 2012;152:262-9.
123. Feng J, Anderson K, Liu Y, Singh AK, Ehsan A, Sellke FW. Cyclooxygenase 2 contributes to bradykinin-induced microvascular responses in peripheral arterioles after cardiopulmonary bypass. J Surg Res 2017;218:246-52.
124. Dharmashankar K, Widlansky ME. Vascular endothelial function and hypertension: insights and directions. Curr Hypertens Rep 2010;12:448-55.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Feng J, Kant S, Sellke FW. Microvascular dysfunction following cardioplegic arrest and cardiopulmonary bypass. Vessel Plus 2021;5:30. http://dx.doi.org/10.20517/2574-1209.2021.57
AMA Style
Feng J, Kant S, Sellke FW. Microvascular dysfunction following cardioplegic arrest and cardiopulmonary bypass. Vessel Plus. 2021; 5: 30. http://dx.doi.org/10.20517/2574-1209.2021.57
Chicago/Turabian Style
Feng, Jun, Shawn Kant, Frank W. Sellke. 2021. "Microvascular dysfunction following cardioplegic arrest and cardiopulmonary bypass" Vessel Plus. 5: 30. http://dx.doi.org/10.20517/2574-1209.2021.57
ACS Style
Feng, J.; Kant S.; Sellke FW. Microvascular dysfunction following cardioplegic arrest and cardiopulmonary bypass. Vessel Plus. 2021, 5, 30. http://dx.doi.org/10.20517/2574-1209.2021.57
About This Article
Copyright

Data & Comments
Data



 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 16
likes
Like This Article 16
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.