
Non-autonomous effects of CCM genes loss
 0
0
Abstract
Cerebral cavernous malformation (CCM) is a rare disease of proven genetic origin characterized by vascular lesions affecting capillaries and small vessels of the central nervous system. CCM lesions occur in a range of different phenotypes, including wide differences in lesion number, size, and susceptibility to intracerebral hemorrhage. CCM lesion genesis requires loss of function of any of three genes, namely KRIT1 (CCM1),
Keywords
INTRODUCTION
Cerebral cavernous malformation (CCM), is a rare disease characterized by “mulberry-like” vascular lesions present in the brain (caverns) with abnormal ultrastructural features of vessels. The thin vessel wall is enlarged and leaky and composed of amorphous material lacking endothelial support and devoid of normal and intact tight junctions and vessel structural components, such as pericytes and astrocyte foot processes[1].
CCM lesions are predominantly found in the central nervous system (CNS) but can be present also in the retina, skin, and liver. In the brain, CCM lesions can occur as single or multiple (even hundreds) lesions, with sizes varying from a few millimeters to a few centimeters, depending on the pathogenesis[2].
Despite the high prevalence of CCM lesions, the development of clinical symptoms is extremely variable, and approximately only 30% of affected people show headaches, focal neurological deficits (25%), seizures (50%), and symptomatic hemorrhage (25%)[3]. Symptoms may occur at any age, although the typical age of onset is between the second and fifth decade of life and without sex predominance, even though the available data are conflicting. Moreover, the majority of CCM lesions remain clinically and biologically quiescent during the most of the host’s lifetime[4-6]. To date, diagnosis is made only by magnetic resonance imaging (MRI), and it is mainly possible only when lesions become symptomatic[3]. The most commonly used classification of CCMs in the literature was proposed by Zabramski et al.[7]. Individual cavernous malformations are divided into four categories on the basis of pathological correlations and magnetic resonance signal characteristics: Type I, characterized by the presence of subacute hemorrhage; Type II, with loculated areas of hemorrhage and thrombosis of varying age; Type III, with chronic areas of hemorrhage; and Type IV, with small multiple punctate microhemorrhagies[7].
CCM is a disease of proven genetic origin (OMIM 116860) that may arise sporadically or can be inherited as an autosomal dominant condition, with incomplete penetrance and variable expressivity. The sporadic form accounts for up to 80% of cases and is characterized by a lack of family history of the disease and the presence of a single lesion on MRI. The familial form accounts for at least 20% of cases and is generally characterized by the presence of multiple CCM lesions in the brain associated with cutaneous and retinal vascular lesions in 9% and 5% of familial CCM cases[8]. Human genetic studies have so far identified three genes whose heterozygous loss-of-function mutations are associated with CCM disease: KRIT1 (CCM1), MGC4607 (CCM2), and PDCD10 (CCM3). The mutation rates for each gene range between 53% and 65% for KRIT1/CCM1, 15% and 19% for CCM2, and 10% and 16% for CCM3[9,10]. However, genotype does not entirely explain the large clinical variability of CCM disease, even among family members carrying the same mutation, suggesting that genetic and environmental modifiers can contribute to CCM disease[11]. Despite the differences between families, some genotype-phenotype correlations are delineated. While CCM1 and CCM2 induce similar characteristics with later manifestation, mutations on CCM3 drive an early age, aggressive phenotype, with higher risk for cerebral hemorrhage and propensity to multiple meningiomas[2].
At the molecular level, CCM gene loss induces the activation of different signaling pathways[12-14]. In particular CCM deficiency have been associated with hyperactivation of the MEKK3-MEK5-ERK5 kinase cascade, that leads to KLF2 and KLF4 upregulation and subsequent altered expression of bone morphogenic protein 6 (BMP6), thrombomodulin (TM), and thrombospondin 1[15-18], with activation of RhoA-ROCK signaling, and with alteration of the junctional signaling including VE-cadherin and β-catenin[19,20]. A significant alteration of reactive oxygen species balance has been also extensively described[13,21-23].
At present, the pharmaceutical therapy available for the treatment of CCM is very restricted and limited to the treatment of clinical symptoms, as headaches, seizures and epilepsy. The clinical approach to CCM disease depends on the position and size of the lesions and its association with hemorrhages or intractable seizures. When possible, the microsurgical resection of lesions is the eligible route[11,24]. Therefore, the development of new pharmacological strategies is necessary to prevent lesion formation and for treating patients with severe inoperable disease or with multiple lesions. The knowledge of molecular mechanisms at the basis of disease onset appears in this context fundamental to identify new drugs able to inhibit the dysregulation of specific signaling pathways and aberrant immune responses. Recent studies focus on different molecules that are under investigation due to their ability to revert the CCM gene loss effects, such as Vitamin D3 and Tempol (reactive oxygen species inhibitors), atorvastatin and fasudil (Rho/Rock signaling inhibitors), propranolol and bevacizumab [angiogenesis and vascular endothelial growth factor, vascular endothelial growth factor (VEGF), inhibitors], sulindac, LY-364947, and SB-431542 (endothelial-mesenchymal transition inhibitors), TM, and endothelial protein C receptor (EPCR) inhibitors. However, at the present there are no therapies that have demonstrated an effective activity on reducing CCM lesion formation[3,14,24].
CEREBRAL CAVERNOUS MALFORMATIONS ONSET AND DEVELOPMENT
As mentioned, CCM lesions are a consequence of a complete loss-of-function of any of the three CCM genes. In familial cases, mutation is inherited as a constitutional germline mutation with incomplete penetrance, and is associated with a biallelic somatic mutation that occurs in the same cell. In particular, CCM genesis may follow a “two-hit” mechanism in which loss of one of the two alleles (first hit) would be the result of the germline mutation and loss of the second allele (second hit) will occur somatically[25-28].
CCM onset might be explained with the Knudson’s two-hit model of tumor suppressor genes. According to this model, tumors derive from two mutations, one for each of the two alleles of the gene of interest. At the present, several cancer genes exhibit biallelic disruption and truncating point mutations, revealing the validity of the model[29]. According to Knudson hypothesis, vascular lesion formation derives from two mutational events in both hereditary (familial) and non-hereditary (sporadic) cases. In familial CCM the first mutation occurs before conception (germline mutation) and the second after conception (somatic mutation). In sporadic CCM only somatic mutations are present.
However, the mechanisms at the basis of these mutations are unknown and some questions still remain open. It is not completely understood if a single second somatic mutation for familial CCM is sufficient to induce vascular lesion formation, where the mutant cells reside within the cellular architecture of the vascular wall and how this second mutation can promote vascular malformation.
In familial CCM, the identification of the somatic mutation remains technically challenging also by using very sensitive approaches as next-generation sequencing techniques. The expected allele frequency of somatic mutation within vascular lesions is low due to the tissue nature, characterized by a vascular structure with a single monolayer of endothelial cells. However, some reports describe a genetic two-step inactivation of all the three genes involved in CCM onset[25-27,30-32].
In the last three years new experimental evidences, supported by mouse models and sophisticated cellular and molecular biology techniques, sustained the idea that the loss of a CCM gene in a single cell may be responsible for CCM development[30,33,34].
Detter et al.[33], by using a CCM3 KO mouse model, showed for the first time that vascular lesions are composed of both mutated and non-mutated endothelial cells. The authors propose a model in which endothelial cells acquire a somatic mutation promoted by a second hit, and undergo clonal expansion subsequent to the acquisition of a high proliferative capability. This clonal expansion is followed by a massive recruitment of non-mutated endothelial cells that become part of the lesion [Figure 1][33].
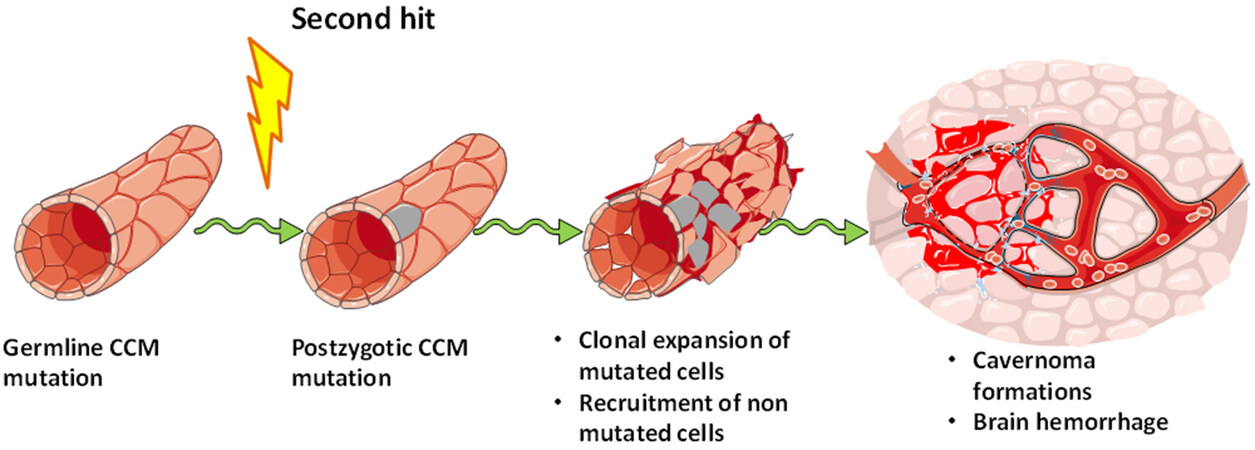
Figure 1. Vascular lesion formation after clonal expansion of mutated endothelial cells. Model of cerebral cavernous malformation (CCM) development in which a second hit promotes a postzygotic CCM mutation that leads to a clonal expansion and the recruitment of wild type endothelial cells into growing vascular lesions. This figure was created using Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com.
These first observations for CCM3 dependent cavernomas have been confirmed by Malinverno et al.[34]. In both mice and humans, the endothelial cells lining the lesion appear as a mosaic, formed by both mutated and non-mutated endothelial cells. Only a very small part of this mosaic is made up of CCM3-/- endothelial cells (about 15% of total endothelial cells of the lesion). Malinverno et al.[34] report interesting data demonstrating that CCM3-/- cells are able to recruit surrounding CCM3+/+ cells and to induce them to undergo endothelial-mesenchymal transition (EndMT) through BMP6. Therefore, an important event in the evolution of the cavernomas appears to be the attraction of CCM3+/+ cells by CCM3-/- cells, which, in turn, strongly contribute to the growth of the malformation[34].
These new findings on CCM3 loss dependent vascular lesions have been confirmed also for CCM1/KRIT1. In the paper by Rath et al.[30], immunohistochemical analysis of samples of a patient with double KRIT1 mutation revealed scattered immunopositivity and immunonegativity of CCM1 staining in endothelial of caverns, while CCM2 and CCM3 were uniformly immunopositive, indicating that the second KRIT1 postzygotic mutation occurs only in a little number of endothelial cell present in the lesion.
In the light of the above observations, it is evident that CCM vascular lesions are not composed of a homogenous layer of mutated endothelial cells, but may be defined as a mosaic of CCM mutated and non-mutated endothelial cells [Figure 1]. These observations could also explain the difficulties in the identification of the second postzygotic mutations, as the reduced number of mutated endothelial cells in vascular lesions imposes the use of extremely sensitive techniques. Moreover, the mechanism by which wild type endothelial cells become incorporated in the growing malformation is unknown even under investigation. The identification of specific signal molecules able to promote the recruitment of wild type endothelial cells into growing lesions may represent a challenge for the identification of new therapeutic strategies.
NON-AUTONOMOUS EFFECTS OF CCM GENE LOSS
The hypothesis that CCM mutated cells become able to recruit non-mutated cells was initially formulated for CCM3 by Louvi et al.[35]. The authors showed that loss of CCM3 in astrocytes induces both cell autonomous and non-autonomous effects. Ccm3-/- astrocytes showed increased proliferation and survival through the activation of Akt signaling, but were also able to induce vascular lesion formation in mouse models, with diffusely dilated and simplified vascular tree, similar to human cavernomas[35]. Similarly,
In this context, we recently demonstrated that Krit1 deficient stromal cells are able to promote the activation of non-mutated endothelial cells by inducing their proliferation and migration[38]. By using Krit1-/- cell models, we showed that Krit1 loss-of-function causes the upregulation of NADPH oxidase isoform 1 (NOX1) and COX-2 in stromal cells and induces activation of inflammatory pathways, which, in turn, promote an enhanced production of proangiogenic factors, including VEGF and Prostaglandin E2 (PGE2). We also showed that conditioned media from Krit1 null mouse fibroblasts induce proliferation, migration, matrix metalloproteinase 2 activation, and VE-cadherin redistribution in wild type human endothelial cells. Consistently, NOX1 and COX-2 inhibition was sufficient to reverse the proangiogenic effects induced by Krit1 loss[38].
These findings allow to hypothesize a model in which endothelial cells, astrocytes, or stromal cells lacking of any of CCM genes induce the activation of non-mutated endothelial cells and the consequent formation of vascular lesions [Figure 2].
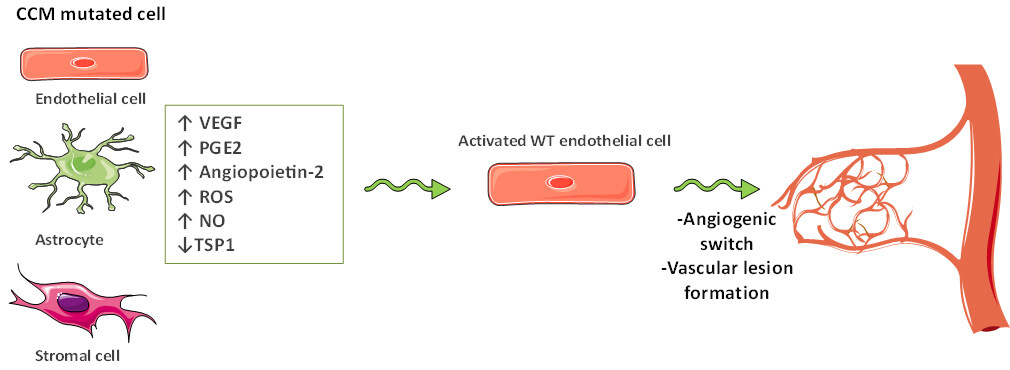
Figure 2. The model by which endothelial cells, astrocytes, or stromal cells lacking one of cerebral cavernous malformations (CCM) genes induce the activation of non-mutated endothelial cells and the consequent formation of vascular lesions. This figure was created using Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com. VEGF: vascular endothelial growth factor; PGE2: prostaglandin E2; ROS: reactive oxygen species; NO: nitric oxide; TSP1: thrombospondin-1.
The non-autonomous effects associated to KRIT1 loss have been described also in the intestine
PROMOTION OF ANGIOGENESIS AFTER CCM GENE LOSS
Loss of CCM genes have been extensively correlated with increased angiogenesis and upregulation of angiogenic factors. In cellular experimental models, endothelial cells silenced for KRIT1 expression increased pseudocapillary and tube formation in 3D assays[40]. Both stromal and endothelial cells depleted in KRIT1 increased VEGF and PGE2 expression and vascular endothelial growth factor receptor (VEGFR) phosphorylation, and reduced the expression of the potent antiangiogenic molecule thrombospondin 1[17,38,41]. Consistently, endothelial cells isolated from human CCM specimens are characterized by increased proliferation and migration and are able to form pseudocapillary networks[42-44]. Likewise, CCM3 deficiency is involved in the angiogenic process through several mechanisms[45]. CCM3 loss is responsible for an increased release of angiopoietin-2, linked to decreased endothelial cell-cell adhesion and pericytes coverage leading to blood vessel disorganization[46]. In addition CCM3 loss promotes increased VEGFR and epidermal growth factor receptor expression in both endothelial and non-endothelial cells[47]. In this scenario, VEGF and PGE2 appear as good candidates to promote the angiogenic switch of quiescent endothelial cells, and VEGF or PGE2 production or signaling cascade inhibition may represent good candidates for CCM therapy[37,38,48].
Another important connection between CCM and angiogenesis is the evidence that CCM gene loss results in impaired DLL-4/NOTCH signaling[40,49,50]. The earliest report on Notch signaling in CCM pathogenesis came from the studies of Whitehead et al.[52] that demonstrated that vascular defects in mice embryos deficient of Krit1 gene resulted from the downregulation of Notch-related genes including Dll4 and Notch4. These data were further confirmed by Wüstehube et al.[40] which reported that CCM1 acts as an anti-angiogenic protein and inhibits endothelial proliferation, migration, and sprouting angiogenesis in human endothelial cells through the induction of DLL4-Notch signaling and the increasing of AKT protein phosphorylation. Consistently, KRIT1 silencing further led to diminished DLL4, HEY1, and HEY2 expression[40].
Moreover, Maddaluno et al.[15], demonstrated, in Ccm1 deficient models, that endothelial cells lining the cavernomas undergo an EndMT induced by a strong activation of the phospho-SMAD-dependent signaling pathway linked to an increased endogenous production of the BMP6 ligand[15]. Endothelial cells that acquire the mesenchymal phenotype are characterized by loss of correct polarization, increased migration rate, and alteration in cell-cell and cell-matrix interactions[52,53]. Loss-of-function of CCM proteins has also been shown to trigger MEKK3 signaling and to increase the expression of the mechanosensitive transcription factors KLF2 and KLF4 that are considered hallmarks of CCM-related EndMT[18,24,54,55]. In addition, it has been recently demonstrated that KLF2 and KLF4 upregulation linked to CCM loss is also responsible for increased levels of anticoagulant endothelial receptors TM and EPCR, as well as of enhanced generation of activated protein C in endothelial cells that are associated to central nervous system hemorrhage in CCM disease[56].
In the light of the above reported data, CCM proteins appear to be key regulators of angiogenesis. The capability to promote endothelial cell proliferation and migration, pseudocapillary formation, and activation of angiogenic signal, may be consistent with the new evidence of the presence of mosaicism in CCM vascular lesions. Loss of one of CCM genes appears sufficient to promote endothelial cell mobilization through autocrine and paracrine mechanisms of action [Figure 2].
CONCLUSIONS
In this manuscript we overviewed the very recent advances in understanding the molecular and cellular mechanisms underlying the onset and progression of CCM disease. In the last few years new experimental evidences proposed a new perspective in the study of CCM and a new model for the CCM lesions formation and development. According to these recent studies vascular lesions appear to be composed by a mosaicism of mutated and non-mutated endothelial cells, in which mutated cells, with high angiogenic features, promote the activation of non-mutated endothelial cells that move from quiescent to active state. CCM mutated cells become able to secrete angiogenic factors and to reduce the production of antiangiogenic factors, thus creating an imbalance between pro- and anti-angiogenic molecules that induces the formation of a new defective vascular network.
Despite the few numbers of papers published so far, the data are robust and very promising. Deeper studies on the mechanisms at the basis of mosaicism observed in CCM will lead to the identification of molecular mediators and signaling pathways that contribute to the recruitment of non-mutated endothelial cells into growing vascular network forming the cavernoma. These findings will permit to identify new targets for the development of novel therapies or for evaluating drug repurposing aimed to prevent CCM onset and progression. The possibility of designing therapeutic approaches aimed both at preventing growth of mutated cells and recruitment of non-mutated cells will represent the new era for the treatment of CCM.
DECLARATIONS
Authors’ contributionsMade substantial contributions to conception and design of the study and performed data analysis and interpretation: Finetti F, Trabalzini L
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by MIUR (Progetto Dipartimento di Eccellenza 2018-2022).
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Wong JH, Awad IA, Kim JH. Ultrastructural pathological features of cerebrovascular malformations: a preliminary report. Neurosurgery 2000;46:1454-9.
2. Padarti A, Zhang J. Recent advances in cerebral cavernous malformation research. Vessel Plus 2018;2:21.
3. Awad IA, Polster SP. Cavernous angiomas: deconstructing a neurosurgical disease. J Neurosurg 2019;131:1-13.
4. Xie MG, Li D, Guo FZ, et al. Brainstem cavernous malformations: surgical indications based on natural history and surgical outcomes. World Neurosurg 2018;110:55-63.
5. Flemming KD, Graff-Radford J, Aakre J, et al. Population-based prevalence of cerebral cavernous malformations in older adults: Mayo Clinic Study of Aging. JAMA Neurol 2017;74:801-5.
6. Flemming KD, Lanzino G. Cerebral cavernous malformation: what a practicing clinician should know. Mayo Clin Proc 2020;95:2005-20.
7. Zabramski JM, Wascher TM, Spetzler RF, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg 1994;80:422-32.
8. Spiegler S, Rath M, Paperlein C, Felbor U. Cerebral cavernous malformations: an update on prevalence, molecular genetic analyses, and genetic counselling. Mol Syndromol 2018;9:60-9.
9. Denier C, Labauge P, Bergametti F, et al. Société Française de Neurochirurgie. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol 2006;60:550-6.
10. Riant F, Cecillon M, Saugier-Veber P, Tournier-Lasserve E. CCM molecular screening in a diagnosis context: novel unclassified variants leading to abnormal splicing and importance of large deletions. Neurogenetics 2013;14:133-41.
11. Akers A, Al-Shahi Salman R, A Awad I, et al. Synopsis of guidelines for the clinical management of cerebral cavernous malformations: consensus recommendations based on systematic literature review by the angioma alliance scientific advisory board clinical experts panel. Neurosurgery 2017;80:665-80.
12. Su VL, Calderwood DA. Signalling through cerebral cavernous malformation protein networks. Open Biol 2020;10:200263.
13. Retta SF, Glading AJ. Oxidative stress and inflammation in cerebral cavernous malformation disease pathogenesis: two sides of the same coin. Int J Biochem Cell Biol 2016;81:254-70.
14. Retta SF, Perrelli A, Trabalzini L, Finetti F. From genes and mechanisms to molecular-targeted therapies: the long climb to the cure of cerebral cavernous malformation (CCM) disease. Methods Mol Biol 2020;2152:3-25.
15. Maddaluno L, Rudini N, Cuttano R, et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013;498:492-6.
16. Zhou Z, Tang AT, Wong WY, et al. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature 2016;532:122-6.
17. Lopez-Ramirez MA, Fonseca G, Zeineddine HA, et al. Thrombospondin1 (TSP1) replacement prevents cerebral cavernous malformations. J Exp Med 2017;214:3331-46.
18. Zhou Z, Rawnsley DR, Goddard LM, et al. The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression. Dev Cell 2015;32:168-80.
19. Glading A, Han J, Stockton RA, Ginsberg MH. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol 2007;179:247-54.
20. Glading AJ, Ginsberg MH. Rap1 and its effector KRIT1/CCM1 regulate beta-catenin signaling. Dis Model Mech 2010;3:73-83.
21. Antognelli C, Trapani E, Delle Monache S, et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free Radic Biol Med 2018;115:202-18.
22. Goitre L, DiStefano PV, Moglia A, et al. Up-regulation of NADPH oxidase-mediated redox signaling contributes to the loss of barrier function in KRIT1 deficient endothelium. Sci Rep 2017;7:8296.
23. Goitre L, De Luca E, Braggion S, et al. KRIT1 loss of function causes a ROS-dependent upregulation of c-Jun. Free Radic Biol Med 2014;68:134-47.
24. Chohan MO, Marchiò S, Morrison LA, et al. Emerging pharmacologic targets in cerebral cavernous malformation and potential strategies to alter the natural history of a difficult disease: a Review. JAMA Neurol 2019;76:492-500.
25. Gault J, Shenkar R, Recksiek P, Awad IA. Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke 2005;36:872-4.
26. Akers AL, Johnson E, Steinberg GK, Zabramski JM, Marchuk DA. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): evidence for a two-hit mechanism of CCM pathogenesis. Hum Mol Genet 2009;18:919-30.
27. McDonald DA, Shi C, Shenkar R, et al. Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum Mol Genet 2014;23:4357-70.
28. Riant F, Bergametti F, Ayrignac X, Boulday G, Tournier-Lasserve E. Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. FEBS J 2010;277:1070-5.
30. Rath M, Pagenstecher A, Hoischen A, Felbor U. Postzygotic mosaicism in cerebral cavernous malformation. J Med Genet 2020;57:212-6.
31. Gault J, Awad IA, Recksiek P, et al. Cerebral cavernous malformations: somatic mutations in vascular endothelial cells. Neurosurgery 2009;65:138-44; discussion 144.
32. Pagenstecher A, Stahl S, Sure U, Felbor U. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet 2009;18:911-8.
33. Detter MR, Snellings DA, Marchuk DA. Cerebral cavernous malformations develop through clonal expansion of mutant endothelial cells. Circ Res 2018;123:1143-51.
34. Malinverno M, Maderna C, Abu Taha A, et al. Endothelial cell clonal expansion in the development of cerebral cavernous malformations. Nat Commun 2019;10:2761.
35. Louvi A, Chen L, Two AM, Zhang H, Min W, Günel M. Loss of cerebral cavernous malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology. Proc Natl Acad Sci U S A 2011;108:3737-42.
36. Wang K, Zhang H, He Y, et al. Mural cell-specific deletion of cerebral cavernous malformation 3 in the brain induces cerebral cavernous malformations. Arterioscler Thromb Vasc Biol 2020;40:2171-86.
37. Lopez-Ramirez MA, Soliman SI, Hale P, et al. Non cell-autonomous effect of astrocytes on cerebral cavernous malformations. BioRxiv 2021; doi: 10.1101/2021.01.29.428891.
38. Finetti F, Schiavo I, Ercoli J, et al. KRIT1 loss-mediated upregulation of NOX1 in stromal cells promotes paracrine pro-angiogenic responses. Cell Signal 2020;68:109527.
39. Chapman EM, Lant B, Ohashi Y, et al. A conserved CCM complex promotes apoptosis non-autonomously by regulating zinc homeostasis. Nat Commun 2019;10:1791.
40. Wüstehube J, Bartol A, Liebler SS, et al. Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc Natl Acad Sci U S A 2010;107:12640-5.
41. DiStefano PV, Kuebel JM, Sarelius IH, Glading AJ. KRIT1 protein depletion modifies endothelial cell behavior via increased vascular endothelial growth factor (VEGF) signaling. J Biol Chem 2014;289:33054-65.
42. Baev NI, Awad IA. Endothelial cell culture from human cerebral cavernous malformations. Stroke 1998;29:2426-34.
43. Zhao Y, Tan YZ, Zhou LF, Wang HJ, Mao Y. Morphological observation and in vitro angiogenesis assay of endothelial cells isolated from human cerebral cavernous malformations. Stroke 2007;38:1313-9.
44. Glading AJ, Finetti F, Trabalzini L. Disease models in cerebral cavernous malformations. Drug Discov Today Dis Model 2020;31:21-9.
45. Wang K, Zhou HJ, Wang M. CCM3 and cerebral cavernous malformation disease. Stroke Vasc Neurol 2019;4:67-70.
46. Jenny Zhou H, Qin L, Zhang H, et al. Endothelial exocytosis of angiopoietin-2 resulting from CCM3 deficiency contributes to cerebral cavernous malformation. Nat Med 2016;22:1033-42.
47. Sartages M, Floridia E, García-Colomer M, et al. High levels of receptor tyrosine kinases in ccm3-deficient cells increase their susceptibility to tyrosine kinase inhibition. Biomedicines 2020;8:624.
48. DiStefano PV, Glading AJ. VEGF signalling enhances lesion burden in KRIT1 deficient mice. J Cell Mol Med 2020;24:632-9.
49. Schulz GB, Wieland E, Wüstehube-Lausch J, et al. Cerebral cavernous malformation-1 protein controls DLL4-Notch3 signaling between the endothelium and pericytes. Stroke 2015;46:1337-43.
50. You C, Zhao K, Dammann P, et al. EphB4 forward signalling mediates angiogenesis caused by CCM3/PDCD10-ablation. J Cell Mol Med 2017;21:1848-58.
51. Whitehead KJ, Plummer NW, Adams JA, Marchuk DA, Li DY. Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development 2004;131:1437-48.
52. Bravi L, Malinverno M, Pisati F, et al. Endothelial cells lining sporadic cerebral cavernous malformation cavernomas undergo endothelial-to-mesenchymal transition. Stroke 2016;47:886-90.
53. Dejana E, Hirschi KK, Simons M. The molecular basis of endothelial cell plasticity. Nat Commun 2017;8:14361.
54. Fisher OS, Liu W, Zhang R, et al. Structural basis for the disruption of the cerebral cavernous malformations 2 (CCM2) interaction with Krev interaction trapped 1 (KRIT1) by disease-associated mutations. J Biol Chem 2015;290:2842-53.
55. Cullere X, Plovie E, Bennett PM, MacRae CA, Mayadas TN. The cerebral cavernous malformation proteins CCM2L and CCM2 prevent the activation of the MAP kinase MEKK3. Proc Natl Acad Sci U S A 2015;112:14284-9.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Finetti F, Trabalzini L. Non-autonomous effects of CCM genes loss. Vessel Plus 2021;5:29. http://dx.doi.org/10.20517/2574-1209.2021.49
AMA Style
Finetti F, Trabalzini L. Non-autonomous effects of CCM genes loss. Vessel Plus. 2021; 5: 29. http://dx.doi.org/10.20517/2574-1209.2021.49
Chicago/Turabian Style
Finetti, Federica, Lorenza Trabalzini. 2021. "Non-autonomous effects of CCM genes loss" Vessel Plus. 5: 29. http://dx.doi.org/10.20517/2574-1209.2021.49
ACS Style
Finetti, F.; Trabalzini L. Non-autonomous effects of CCM genes loss. Vessel Plus. 2021, 5, 29. http://dx.doi.org/10.20517/2574-1209.2021.49
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 4 clicks
Cite This Article 4 clicks

 Like This Article 6
likes
Like This Article 6
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.