
The role of uric acid in renal damage - a history of inflammatory pathways and vascular remodeling

Abstract
The association of hyperuricemia with cardiovascular risk, hypertension, atherosclerosis, metabolic syndrome, mortality, and chronic kidney disease has been largely described in clinical studies. Several pathogenetic mechanisms explaining uric acid mediated renal damage have been hypothesized, including crystal deposition, oxidative stress, arteriolosclerosis, and glomerular hypertension. Currently, two explanations for hyperuricemia-induced renal injury are the most widely accepted. Firstly, the fact that uric acid is recognized by receptors involved in the innate immune response as a dangerous molecule appears to be a powerful trigger for the inflammatory cascade, which ultimately lead to renal fibrosis. Secondly, serum uric acid has been demonstrated to be implicated in the renin-angiotensin system activation and nitric oxide synthesis inhibition, which promote endothelial dysfunction and proliferation of vascular smooth muscle cells, resulting in glomerulosclerosis and interstitial fibrosis. In this review, we focus on experimental data demonstrating pathophysiological mechanisms linking uric acid to inflammation and oxidative stress, which contribute to the development and progression of renal injury. In addition, we describe endothelial and vascular dysfunction crucial playmakers in kidney impairment induced by uric acid.
Keywords
INTRODUCTION
Uric acid (UA) is the end product of nucleic acid metabolism and it is synthesized mostly in the liver, intestines, muscles, kidneys and the vascular endothelium. Due to UA renal excretion, hyperuricemia has always been closely related to chronic kidney disease (CKD). Glomerular filtration rate is undoubtedly one of the main determinants of serum UA levels and the impairment of renal function is the first target organ damage determined by UA[1]. Kidney function impairment and hyperuricemia are well known to increase cardiovascular risk[1-4], even though the independent pathogenic contribution of each one of these variables on increasing cardiovascular risk and mortality is a current research topic[5]. Furthermore, hyperuricemia is closely related to metabolic syndrome, obesity and diabetes which are the natural consequence of insulin resistance and common risk factors for CKD. Due to the fact that insulin reduce urinary urate excretion by tubular reabsorption, CKD and insulin resistance are both bi-directionally related to serum uric acid, with one variable possibly contributing to influence the other one[6].
These complex interactions motivated the search for the possible pathogenetic mechanism by which increased UA levels could cause tissue damage and therefore contribute to the development of hypertension, insulin resistance, CKD, end-stage renal disease, cardiovascular events, and mortality. To the present day, a lot of epidemiological data have associated hyperuricemia with closely related vascular diseases such as kidney disease[2], showing a strong association between higher circulating UA levels and CKD occurrence and progression[7-9]. The current debate on the unclear benefit of treating hyperuricemia to slow CKD increasingly inspires the study of the molecular mechanisms underlying hyperuricemia-mediated organ damage.
Several experimental studies defined potential pathways linking UA to CKD lesions. Mechanisms include, inflammation, oxidative stress, activation of the renin-angiotensin aldosterone system (RAAS), endothelial dysfunction, proliferation of vascular smooth muscle cells (VSMCs), resulting in glomerulosclerosis and interstitial fibrosis[10]. Interestingly, it has been found that UA is recognized by receptors involved in the innate immune response as a dangerous molecule, which acts as a trigger for the inflammatory cascade and therefore has been classified as one of the damage associated molecular patterns (DAMPs). Moreover, oxidative stress increases cytokine release and adipokine synthesis as well as inflammation, all elements that have been suggested as important factors in mediating kidney damage[10]. In the kidney indeed, cytokines induce expression of reactive oxygen/nitrogen species[11], bioactive lipids[12] and adhesion molecules[13] promoting aberrant matrix metabolism[14] and proliferation of resident cells[15].
Furthermore, there is much evidence of the role of UA in aberrant changes in vascular properties. These include endothelial dysfunction[16], promotion of VSMCs proliferation[17], and induction of vasoconstrictive mediators such as endothelin-1 (ET-1) and angiotensin II (Ang II)[18,19]. Experimental evidences suggest a complex but potentially direct, causal role of UA in the pathogenesis of atherosclerosis[7].
In this review, we focus on experimental data demonstrating pathophysiological mechanisms linking UA to the processes leading to vascular and systemic inflammatory response, thus contributing to the development and progression of renal injury. Cellular and hemodynamics effects of UA explaining the experimental, clinical, and epidemiologic relationship that has been described with CKD[6,20,21] are depicted in Figure 1.
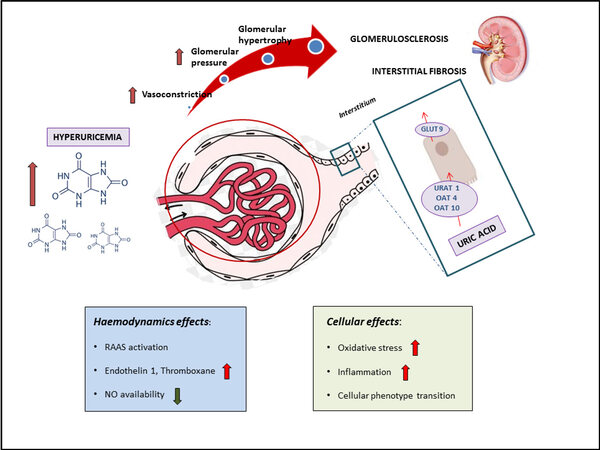
Figure 1. Pathological effects of hyperuricemia on kidney structure and function. Pathophysiology of uric acid - mediated kidney damage. Uric acid enters renal tubular cells through a specific transporter, urate transporter 1 (URAT-1), and two generic transporters, organic anion transporter 4 (OAT-4) and OAT-10. On the basolateral site, the glucose transporter 9 (GLUT-9) is the principal transporter responsible for extrusion of uric acid into circulation. Hyperuricemia might cause hemodynamic effects including increased activity of the RAAS, increased production and activity of vasoconstrictors, such as ET-1, Ang II and thromboxane, and impairment in nitric oxide (NO) availability. These changes lead to impaired endothelium-dependent relaxation and endothelial dysfunction, with negative consequence on kidney structure and function. Moreover, uric acid has been demonstrated to have cellular effects inducing oxidative stress, inflammation and cellular phenotype transition, contributing to glomerulosclerosis and interstitial fibrosis. RAAS: renin angiotensin aldosterone system; NO: nitric oxide; GLUT: glucose transporter; URAT: urate transporter; OAT: organic anion transporter.
HYPERURICEMIA-INDUCED RENAL INJURY
In the first part of this review, we briefly describe the detrimental effects of UA acting as a DAMP and increasing oxidative stress and promoting inflammation. In the second part, we focus on signaling pathways by which UA could cause the renal vascular remodeling.
The innate immune response
Besides the glomerular and tubular changes directly induced by UA, immune cells populating the kidney recognizes it as a dangerous molecule, and in turn have a direct detrimental effect on renal cells[22]. The main known players of UA immune-recognition are the Nod-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome and toll-like receptors (TLRs), both expressed by renal proximal tubular cells[23].
Inflammasome
Hyperuricemia leads to the formation and deposition of monosodium urate (MSU) crystals, a remarkable event in the pathology of hyperuricemic-related diseases[24]. Several in vitro studies demonstrate an inflammatory-related response triggered by MSU irrespective of the cell type, although not all the intracellular pathways have yet been revealed[23]. MSU crystals are able to turn on human primary macrophages to secrete the lysosomal protease cathepsin, proinflammatory cytokines, such as interleukin (IL)-1β, IL-18 [Figure 2], and interferon through the Src/Pyk2/PI3K signaling pathway[25]. An important member of NLRP3 plays a key role in this pathway.
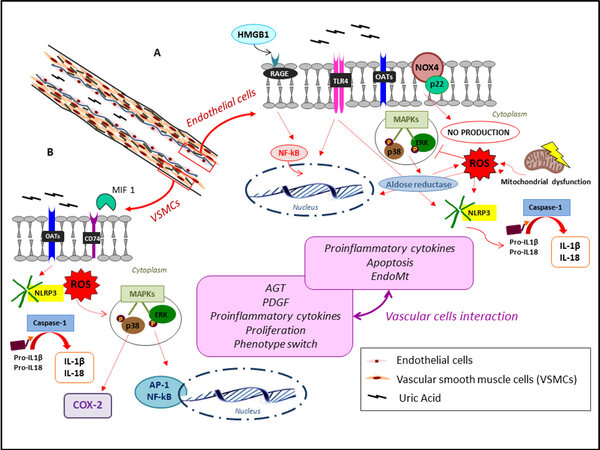
Figure 2. Pathways for uric acid-mediated endothelial (A) and vascular smooth muscle cell (B) damage. In endothelial cells, high uric acid levels stimulate the RAGE signaling pathway and activates NF-κB. This process conduces to the extracellular release of HMGB1 in endothelial cells, and its interaction with RAGE due to its high affinity contributes to the amplification of the inflammatory response, finally inducing endothelial dysfunction. In addition, intracellular uric acid causes P38 and extracellular signal-regulated kinase (ERK) 42/44 MAPK phosphorylation, increasing aldose reductase expression and inducing NOX4 over expression and ROS production. Moreover, uric acid contributes to a pro-inflammatory state, mediated by TLR-4 with NOX4 up-regulation, NLRP3 activation and interleukin production, promoting cellular switching and apoptosis. The ROS produced by xanthine oxidase are an important messenger inducing inflammation and signal transduction, such as mitochondrial dysfunction, leading to apoptosis, increase in proinflammatory cytokines and phenotype transition (A). In VSMCs, uric acid induces proinflammatory cytokine production, apoptosis, and endothelial-mesenchymal transition by several pathways. Uric acid enters the vascular smooth muscle cell where it alters intracellular redox, activates mitogen activated protein kinases (Erk1/2 and p38), COX-2, and nuclear transcription factors (NFκB and AP-1), leading to synthesis of cytokines and PDGF, as well as proliferation and phenotype transition of these cells. The ROS increase and NLP3 activation have similar effects as in endothelial cells. Finally, it has been demonstrated UA-mediated up-regulation of macrophage MIF protein, a cytokine playing inflammatory response induced in VSMCs by oxidized low-density lipoproteins and Ang II during atherogenesis. The two pink boxes summarize the UA-mediated effects on the two cell types, and their interaction brings up an interesting and new aspect of the research, as described in the text (B). AGT: Angiotensinogen; AP-1: activator protein-1; COX-2: inducible cyclo-oxygenase; HMGB1: high-mobility group protein-1; IL: interleukin; MAPK: mitogen-activated protein kinase; MIF: migration inhibitory factor; NLRP3: Nod-like receptor pyrin domain-containing protein 3; NOX4: NADPH oxidase 4; ROS: reactive oxygen species; OAT: organic anion transporter; RAGE: receptor for advanced glycation end products; TLR4: toll-like receptor 4; VSMC: vascular smooth muscle cell.
NLRP3 is currently the most well-recognized Nod-like receptor and the most widely studied inflammasome in the field of kidney diseases[26]. The activation of NLRP3 inflammasome requires a priming signal to induce transcription of both NLRP3 and pro-IL-1β, and a second signal to prompt oligomerization of the inflammasome. Several ligands can induce NLRP3 priming, including the TLR 2 ligand Pam3CSK4 (Pam3) and the TLR4 ligand LPS through activation of nuclear factor (NF)-κB[27]. After endocytosis into macrophages, lysosomes attempt to degrade MSU crystal without success. This leads to the lysosomal membrane rupture and lysosomal cathepsins release into the cytoplasm, leading to the activation of the inflammasome[28]. The report by Braga et al.[22] demonstrate that soluble UA activates NLRP3 inflammasome and induces IL-1β release, cellular redox state changes and mitochondrial changes in macrophages. The subsequent transformation of pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, respectively, involves the entire cascade determining sterile inflammation[29,30] and amplifying downstream inflammatory signals[31].
Moreover, MSU crystals promote macrophages to secrete transforming growth factor beta-1 (TGF-β1) through mediation of the metastatic tumor antigen 1/transglutaminase 2 (MTA1/TG2) signaling pathway[32]. TGF-β1 is a strong profibrotic cytokine, and aberrant TGF-β1 derived from MSU crystal-induced macrophages, together with above mechanisms, may promote renal fibrosis, as evidenced in in vitro[33-35] and in vivo[36,37] studies. It has been demonstrated that these extensive biological activities can promote inflammation and dysfunctions in cell metabolism, contributing to the loss of integrity of the glomerular filtration barrier, as outlined in Figure 2. Interestingly, this paves the way for future therapeutic strategies contrasting renal fibrosis based on blocking serum UA internalization or inhibiting its recognition by phagocytes[22,38].
Toll-ike eceptor 4
MSU crystal can be recognized in the extracellular fluid by pattern recognition receptors (e.g., TLRs) expressed on antigen-presenting cells, such as macrophages and tubule epithelial cells as one of the DAMPs which activate immune and inflammatory responses[39,40]. Accumulating evidence demonstrated that TLR4 and other innate immunity-related components (e.g., NLP3, caspase-1, and IL-1β) are essential in the development of UA-mediated inflammation[41], but the mechanisms underlying this pathway still remain largely unclear.
An in vitro study by Xiao et al.[42] showed that soluble UA enhances NLP3 expression, caspase-1 activation, IL-1β and intracellular adhesion molecule (ICAM)-1 production in the human primary renal proximal tubular epithelial cells (PTECs) in a TLR4-dependent pathway.
These processes were also proven in mesangial cells[43,44] and confirmed by further studies[43,45]. Milanesi et al.[46] documented for the first time the additive effect of UA and Ang II in the stimulation of proinflammatory patterns mediated by TLR4 in PETCs.
The biological activity of TLR4 as a key signal molecule in the immuno-inflammatory network pathway has been proven by in vivo studies in mice, suggesting new therapeutic approaches to improve hyperuricemia-mediated immuno-inflammatory renal damage[47,48]. In fact, TLR4 inhibition has proven to reduce soluble UA levels[49,50] and to reduce the severity or slow the progression of the kidney damage[51-53].
Oxidative stress
Oxidative stress is a phenomenon caused by the imbalance between the formation and the removal of free radicals. The most effective free radicals are derived from molecular oxygen, such as superoxide anion (O2) hydrogen peroxide (H2O2), peroxyl radical (ROO) and the very reactive hydroxyl radical (OH) termed ROS, generally considered to be toxic to cells[54]. Many experimental data have suggested a possible role for high hyperuricemia in inducing endothelial dysfunction, and particularly impaired NO bioavailability[55].
Recent studies report that the oxidative stress due to high UA levels directly caused kidney damage and progression of CKD[56]. The mechanisms by which this process takes way continues to be extensively investigated. Hyperuricemia promotes ROS generation and increases oxidative stress inhibiting NO synthesis in several cell types, including mesangial cells, adipocytes, PTECs, and VSMCs[57-59]. This ROS amount determine proliferation, extracellular matrix deposition, and apoptosis[60]. Nevertheless, whether UA contributes to oxidative stress by other specific pathways is still a matter of debate.
The role of urate as a free radical scavenger contributing more than 50% of plasma antioxidant activity has long been recognized by researchers[61]. On the contrary, the description of a redox-dependent effects of UA in PETCs cells primarily produced by NADPH oxidases[62] offers a possible explanation for the paradox by which urate drives oxidative stress when internalized in cells. This last effect may explain the renal protective effects that xantino-oxidase inhibitors showed in some retrospective and randomized studies[63-66]. Moreover, UA proved to contribute to the activation of pro-inflammatory pathways[62,67], as well as the above-mentioned NLP3 inflammasome. This results decrease NO bioavailability, promoting apoptosis of endothelial cells which is part of the so called endothelial dysfunction[68,69].
Mitochondrial damage
Another research hotspot in recent years has become the mitochondrial damage caused by oxidative stress which probably represents the first stimulus leading to the renal tubular epithelial cell apoptosis[70]. One of the main ROS source is NOX4, a member of NADPH oxidase family. It is highly expressed in the kidney and less in VSMCs and endothelial cells. Some studies have demonstrated the involvement in renal tubular cell apoptosis induced by UA[60,71] and its role in atherosclerosis process is well known[72]. The mitochondria constitute the basis of cell energy metabolism, being the primary place where oxidative phosphorylation occurs. Mitochondrial aerobic activity produces a large amount of adenosine triphosphate (ATP), but also of ROS. When cells are in redox conditions in response to environmental stimuli, such as UA increase, the ROS products exceed the amount that can be cleared. Consequently, the mitochondrial membrane lipid peroxidation occurs, reducing the membrane fluidity and swelling of the mitochondria. This leads to the loss of select permeability and therefore to the mitochondrial dysfunction and cell apoptosis[73].
In this regard, it has been found that UA-induced endothelial dysfunction is associated with mitochondrial dysfunction and reduced ATP generation and, lastly, with apoptosis[73,74]. Molecules such as, alpha lipoic acid, that acts as cofactor in mitochondrial dehydrogenase reactions and with antioxidant properties, improve mitochondrial damage and apoptosis stimulated by UA in endothelial cells, through the activation of protein kinase B (Akt/PKB) signaling[75].
Examining rats with a nephropathy induced by UA, investigators found that the urinary protein, oxidative stress index, and the expression of apoptosis proteins, significantly improved in the group treated with glutathione (serving as an antioxidant)[70,76], creating new insight for treatment development.
Vascular system remodeling
As a matter of fact, microvascular renal lesions are associated with the kidney damage progression by impairing the autoregulatory response to blood pressure and by reducing the glomerular blood flow inducing ischemia. In humans, it has been reported that HU is associated with renal microvascular damage, increased renal resistive index, afferent vasoconstriction in healthy subjects, CKD and hypertensive patients[77-80].
Blood vessels are composed of three concentric layers: the intimal layer, composed of a single concentric coat of endothelial cells; the media, composed of smooth muscle cells; and the adventitia, constituted by a complex of extracellular matrix, fibroblasts, and nerve cells.
The glomerulus is a tuft of capillaries lined by endothelial cells and with smooth muscle cells in their wall. These two primary cell types provide a unique and essential contribution to vessel function. Accumulating evidences suggest that increased serum UA levels are associated with vascular cell dysfunction contributing to the development of vascular stiffness[81] and CKD onset/progression[7]. This may reflect the ability of UA firstly to induce the renin-angiotensin system (RAS) activation contributing to atherosclerosis development, and secondly to cause vascular cell changes resulting in endothelial dysfunction, VSMCs proliferation and phenotype switch. All these processes are pivotal events in vascular system remodeling, and therefore determinants of kidney injury.
RAS activation and atherosclerosis
The central role of the RAS in kidney injury is evidenced by the beneficial effects of the RAS blockade in kidney disease[82]. Ang II is known to be a key player in the pathogenesis of metabolic syndrome, which is known to be related to atherosclerosis, hypertension, insulin resistance and CKD progression[83]. Within the kidney, Ang II induces renal microvascular constriction, especially of the efferent arterioles, playing a major role in the regulation of systemic and glomerular blood pressure and therefore of renal function[84]. The RAS contributes to the pathogenesis of kidney injury also by its direct fibrogenic effect on PTECs and VSMCs[85]. In particular, Ang II induces transforming growth factor beta (TGF-β) production, via p38 MAPK activation and JNK/thrombospondin-1 signaling[85], indirectly upregulates epithelial growth factor receptor in renal proximal tubule further enhancing TGF-β induction[86] and directly upregulates adhesion molecule, mRNA and protein synthesis[87,88] contributing to atherogenesis.
The vascular remodeling is certainly a key player for the development of arteriolosclerosis[89,90]. Recent studies have shown serum UA as an independent risk factor for the presence of arteriolar hyalinosis and intimal thickening of the vessel analyzed in kidney biopsies[91,92]. Moreover, Sánchez-Lozada et al.[69] found that raising the serum UA level could induce oxidative stress with endothelial dysfunction, resulting in the development of both systemic and glomerular hypertension, as well as elevated renal vascular resistance and reduced renal blood flow in turn a powerful activator of RAS.
Several experimental observations raise the possibility of a UA-induced nephropathy mediated by the activation of the intrarenal RAS[85,93,94]. Despite the mechanism still unclear, UA is demonstrated to increase inflammatory cytokines and upregulate tissue RAS in rat adipocytes[95]. Sánchez-Lozada et al.[69] showed in their preclinical study that UA interferes with the interaction between Ang II and its Ang-type 1 receptor leading to the development of hypertension. The first evidence of the direct effect of UA on the RAS activation in humans was found by Perlstein et al.[84] and contributed to a possible explanation of the well documented relationship between hyperuricemia and hypertension[93], glomerular hypertrophy[87], afferent arteriolopathy[85], and interstitial inflammation[93] in in vivo studies. Recent studies demonstrating the association between higher serum UA levels and higher ratio of Ang II to angiotensin (1-7) in preterm adolescents support the idea that UA may contribute to increased blood pressure and vascular injury by suppressing angiotensin (1-7)[96].
Despite the well documented efficacy of ACE inhibitors and angiotensin receptor blockers in slowing the kidney disease progression, the nephro-protective treatment needs to be improved and understanding the relationship between UA and RAS might be of utmost importance for this purpose. The recent discovery that RAS and UA play a somewhat additive role both at the renal tubule and at the endothelial level[46,97] confirms that RAAS inhibition alone cannot be the complete solution for the CKD patients and that drugs active on other metabolic pathways could help improve their prognosis.
Endothelial dysfunction
Hyperuricemia is one of the main factors triggering endothelial dysfunction, a primary mechanism for the development of vascular damage[98-101]. The mechanism of endothelial injury involves oxidative stress leading to redox signal pathway activation, the endothelial-to-mesenchymal transition and cytokine and inflammatory factors activation[102].
Experimental studies have shown that human umbilical vein endothelial cells (HUVECs) express 4 different UA transporters: URATv1, ABCG2, MRP4 and MCT9[103,104]. URATv1 plays an important role in the UA internalization[101] then causing oxidative stress through activation of NADPH oxidase, in which the aldose reductase played a central role[99], and reduction of endothelial NO bioavailability [Figure 2]. Moreover, high UA caused P38 and extracellular signal-regulated kinase (ERK) 42/44 MAPK phosphorylation, increasing aldose reductase expression. This process induces NOX4 and ROS over expression with deleterious effects on endothelial cells[99]. In addition, Li et al.[99] proposed that UA could induce oxidative stress through the protein kinase C pathway. Consequently, eNOS activity and NO production are reduced, endoplasmic reticulum stress is induced, and endothelial cell die by mitochondrial oxidation and apoptosis[57,103].
Yet, another key effect of UA on endothelial cell lies in its pro-inflammatory nature. UA increases C-reactive protein production through the activation of p38 and ERK 44/42 mitogen-activated protein kinases pathways[68] and, in addition, several studies demonstrate that UA induces cytokine and chemokine expression through NF-κB activation[104,105]. In this regard, Zhen et al.[105], demonstrate that NF-κB pathway mediates hyperuricemia-induced endothelium impairment and vascular dysfunction, reducing NO and upregulating IL-6, IL-8 and TNF-α expression. The nuclear protein high-mobility group protein-1 (HMGB1) is a pro-inflammatory cytokine that can interact with the receptor for advanced glycation end products (RAGE). Recently, it has been demonstrated that in endothelial cells, the HMGB1/RAGE signaling pathway contributes to endothelial dysfunction induced by UA[106]. Indeed, when RAGE is blocked with a specific antibody, RAGE, HMGB1, ICAM-1, and VCAM-1 are down regulated as well as the DNA binding activity of NF-κB and the release of IL-6 and TNF-α[105]. By proteome analysis of endothelial cells exposed to UA, Oberbach et al.[107], reported that UA may promote a variety of signaling pathways involved in metabolic processes which showed to be fundamental for endothelial homeostasis. In particular, UA regulates ubiquitin-proteasome system, the major pathway of protein degradation. The ubiquitin-proteasome system has been indicated to contribute to dysfunction of endothelial cells in vascular complications during uremia[108] and in HUVECs exposed to Ang II[109], modulating eNOS expression and the availability of cofactors and proteins involved in eNOS activation. In UA exposed cells, a significant increase of proteasome activity as well as ubiquitin and ubiquitinated proteins are observed, suggesting a key role of UA in ubiquitin proteasome system regulation in endothelial dysfunction[110].
In vitro and in vivo findings suggest that UA contributes to endothelial dysfunction by inducing antiproliferative effects on endothelium and impairing NO production. Several studies reported UA-induced endothelial dysfunction as one of the main mechanisms of kidney disease, as a result of impaired vasodilatation and hemodynamic functions and the potential benefit of urate lowering therapy[110,111].
Phenotype transition of endothelial cells
Phenotype transitions of cells have been regarded as one of the earlier mechanisms of kidney disease. As the induction of epithelial‐to‐mesenchymal transition on tubular renal cells[112] results in uncontrolled and exaggerated production of collagen and other extracellular matrix proteins leading to renal fibrosis[113,114], the endothelial‐to‐mesenchymal transition (EndoMT) has been identified as an emerging mechanism of vascular and renal disease, mainly driven by oxidative stress[115,116]. By this process, endothelial cells lose their characteristics (polarity, adhesion, CD31 and eNOS expression) and acquire mesenchymal traits such as a spindle-shape, migratory properties, alpha-smooth muscle actin (α-SMA), fibroblast-specific protein 1 and vimentin expression[117].
Novel findings suggest that UA, increasing NOX activity and, consequently, ROS levels, induces EndoMT in HUVECs and in an animal model of hyperuricemia[118]. Pretreatment with probenecid and antioxidants blunts EndoMT, whereas in hyperuricemic rats, allopurinol partially reverses renal altered expression of vascular endothelial cadherin and α-SMA induced by UA[119].
Ko et al.[118] performed in vitro and in vivo experiment showing as UA induces EndoMT and glycocalyx shedding in cultured vascular endothelial cells. The literature reports as the endothelium is the first line defense against injury[120]. In particular, the loss of the glycocalyx, a gel-like structure formed by proteoglycan core proteins, glycosaminoglycan chains, sialoglycoproteins and adsorbed plasma proteins has been shown to be one of the earliest signs of endothelial injury. Its loss contributes to vascular permeability increase, intravascular thrombosis, loss of NO bioavailability and oxidative stress implicated in several diseases, including kidney involvement[120,121].
As well as an UA-mediated damage mechanism, the detection of glycocalyx shedding is a direct visualization and measurement technique for changes of endothelial cells, being a help for future research on hyperuricemia-induced vascular damage[122].
Specific triggers may induce different levels of EndMT, depending on the underlying cause of renal disease and it could be important to address whether inhibition of EndMT might be a potential therapeutic strategy against renal injury[115].
Vascular smooth muscle cells inflammation, proliferation, and phenotype transition
UA is an independent risk factor for vascular inflammation and remodeling in patients with hypertension or atherosclerosis[123], and this inspired investigators to better understand the role and mechanisms of UA toxicity in VSMCs. One of the first clear demonstration that UA induces arteriolar damage was the finding of expansion of the VSMCs and narrowing of the lumina of the afferent arterioles in hyperuricemic, hypertensive rats[85]. These damages have been shown to be induced by UA per se and not by hypertension because they developed in hyperuricaemic rats with blood pressure well controlled by hydrochlorothiazide and were prevented by allopurinol[85].
In vitro studies have elucidated the possible mechanism of UA-mediated arteriolosclerosis firstly through the demonstration of the presence of urate-transport channel URAT1 in human smooth muscle cells[124]. Moreover, there is long-time evidence that soluble UA can induce VSMC inflammatory response increasing cytokine expression in the vessel wall[125], proliferation[13], and VSCM transition from a contractile state to a secretory state[126].
UA has also been shown to possess proliferative and pro-inflammatory abilities, such as to activate intracellular protein kinases (p38 and Erk 1/2) and activator protein-1 (AP-1). Moreover, UA directly affects VSMC by blocking NO release, inhibiting endothelial proliferation and stimulating C-reactive protein production[16,85,125]. Furthermore, UA has proven to induce VSMC proliferation through activation of specific mitogen-activated protein kinases, nuclear transcription factors (e.g., NF-κB). Thus, the VSMCs produce growth factors (e.g., platelet-derived growth factor PDGF), vasoconstrictive substances like Ang II and thromboxane (TXA2), immune-mediators, and proinflammatory molecules such as C-reactive protein and monocyte chemoattractant protein 1[127].
Kang et al.[128] demonstrated a new mechanism involving inducible cyclo-oxygenase (COX-2) by which UA may directly stimulate the VSMC proliferation. The uthors found de novo expression of COX-2 mRNA by rat aortic VSMC after incubation with UA, and the prevention of UA mediated proliferation incubating these cells with either a COX-2 inhibitor or with a TXA2 receptor inhibitor. Moreover, COX-2 was also shown to be expressed de novo in the preglomerular rat’s vessels, and its expression correlated both with the serum UA and with VSMC proliferation degree[128].
A more recent study from Kırça et al.[129] showed that soluble UA induces proliferative pathways in rat VSMCs along with activation of p38 MAPK, p44/42 MAPK, and PDGF receptor in a time and concentration-dependent manner. Interestingly, inhibitory effects of losartan on p38 and p44/42 MAPK activation were also demonstrated in this study, providing a direct proof of p38 and p44/42 MAPK inhibition by reducing UA internalization thorough losartan blockade of URAT1[129].
With the aim to better understand UA-induced vascular injury mechanisms, Fu et al.[127] treated mice with UA injection and found up-regulation of macrophage migration inhibitory factor (MIF) protein which is a cytokine playing inflammatory response induced in VSMCs by oxidized low-density lipoproteins and Ang II during atherogenesis[127]. Moreover, MIF inhibition alleviated UA-induced vascular inflammation and de-differentiation of VSMCs[130] suggesting that this would be a potential therapeutic approach against vascular injury following UA exposition.
The UA-mediated molecular and cellular pathways involved in endothelial and vascular cells dysfunction are summarized in Figure 2.
Endothelial cell/smooth muscle cell interactions
Signaling between endothelial cells and smooth muscle cells is essential for maintaining tone in mature vessels. Their interaction is critical during development, and for repair and remodeling associated with blood vessel growth. Recently it has been depicted the pathways these cells utilize to communicate and how disruptions in these pathways contribute to the organ damage[131].
The individual functions of endothelial cells and VSMCs are dependent on proper crosstalk between these cell types, which begin during embryogenesis[132]. This process is regulated by growth factors and tissue hypoxia, two elements showed to be involved in UA mediated injury.
Technically, these signaling pathways can be divided into two categories: those mediated by soluble or secreted molecule, and those requiring direct physical contact between the two cell types. As regards the diffusible signaling, both in vitro and in vivo models have demonstrated the role of PDGF signaling in endothelial smooth muscle cell interactions, in particular devoted to smooth muscle recruitment and proliferation, and blood vessel maintenance[133]. Moreover, there is clear evidence that the TGF-β signaling is important for the communication between vascular cells, being receptor-ligand combinations expressed on both endothelial and smooth muscle cells[134].
In several tissues, UA has been shown to stimulate oxidative stress and cellular damage via modulation of TGF-β[135] and the pivotal role of TGF-β-induced tubule-interstitial fibrosis in the progression of CKD is an active topic of research. The attenuation of TGF-β signaling, for instance under angiotensin receptor blockers treatment, has been proven in CKD, by reducing ligands, receptors, and activators[136].
Impaired availability of NO, potentially UA induced, has been found to play a role in signaling via secreted or diffusible factors. In the adult vasculature, it works as hyperpolarizing agents diffuse to VSMCs to cause vascular relaxation[137] that constitutes an efficient mechanism of communication. Likewise, endothelial cell-released contracting agents like ET-1 and Ang II, two known elements strictly involved in UA mediated cellular pathways, are perceived by smooth muscle cells to increase vascular tone[138]. Endothelial cell-dependent regulation of vascular reactivity represents the best-described example of the importance of endothelial-smooth muscle cell interactions, and therefore also in modulation of vascular tone, a key component of the glomerulus.
The second category is represented by contact-dependent signaling, a greater important issue during development then in adults, because of the presence of significant barriers to physical interactions of endothelial-smooth muscle cells in mature blood vessels. However, evidence that cell contact-dependent developmental mediators play a role in adult blood vessel function is an assumption, showing as it might be controlled by Notch signaling[139] and ephrin proteins family, which are noted to modulate blood pressure.
The last key player regarding interaction between cells compounding vascular system is the already in-depth described endothelial dysfunction. A better understanding of how the endothelial dysfunction directly modifies the crosstalk between the two cell types, and the role that UA takes on within this process could be the next advancement in the fight to cure UA-mediated vascular and organ damage.
The magnitude and importance of UA role in the pathogenesis of organ damage might vary and depend on several factors. In the last 20 years, several clinical studies demonstrated as serum UA is a concomitant of high-risk conditions and a predictor of unfavorable outcomes. As a matter of fact, association of hyperuricemia with metabolic syndrome, kidney function impairment, and its impact on cardiovascular disease and mortality is well established[4,6-10,140]. By a pathophysiological point of view, these data are consistent with the hypothesis that UA induces and perpetuates reno-vascular injury, leading to a progressive vicious cycle of further renal damage and thereafter to an increased cardiovascular and all-cause mortality risk. The mechanisms deemed to be implicated in the progression of UA-mediated renal damage are likely multiple and include changes in molecular and cellular systems leading to a wide range of injuries in several tissues, vessels, and organs having a major impact on global health.
CONCLUSION
UA induces immune system activation and alters the characteristics of resident kidney cells, such as tubular epithelial, mesangial, endothelial, and vascular smooth muscle cells, toward a proinflammatory and profibrotic state[141]. Both the hemodynamic and structural changes have been described as key players in UA-induced kidney disease. These findings have led to an increased awareness of UA as a potential and modifiable risk factor in kidney disease, even if the effects of urate lowering therapy are not conclusive.
To the present, UA is considered as a culprit and not only as an innocent bystander in hypertension and progression of renal disease. For this reason, the not univocal demonstration of a benefic effect of urate lowering therapy has to be read as a reflection of incomplete knowledge of UA pathogenetic mechanisms. So, in addition to the need for large clinical trials, more studies are required to better understand the biology of UA.
This review highlights the crucial role of UA in renal cell dysfunction focusing on inflammation and vascular injury, the two main aspects of UA-induced kidney damage. The hope is it will be possible to expand therapeutic strategies by understanding the molecular and cellular processes underlying the occurrence or the progression of CKD induced by hyperuricemia.
DECLARATIONS
Authors’ contributionsDesign and conduct of the study (ER, DV, GL, RP, GG, FV), data interpretation (FC, PE) and manuscript writing (ER, DV, FV): Russo E, Verzola D, Cappadona F, Leoncini G, Garibotto G, Pontremoli R, Viazzi F
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Maloberti A, Maggioni S, Occhi L, et al. Sex-related relationships between uric acid and target organ damage in hypertension. J Clin Hypertens (Greenwich) 2018;20:193-200.
2. Viazzi F, Garneri D, Leoncini G, et al. Serum uric acid and its relationship with metabolic syndrome and cardiovascular risk profile in patients with hypertension: insights from the I-DEMAND study. Nutr Metab Cardiovasc Dis 2014;24:921-7.
3. Bezante GP, Viazzi F, Leoncini G, et al. Coronary flow reserve is impaired in hypertensive patients with subclinical renal damage. Am J Hypertens 2009;22:191-6.
4. Maloberti A, Giannattasio C, Bombelli M, et al. Hyperuricemia and risk of cardiovascular outcomes: The experience of the URRAH (Uric Acid Right for Heart Health) project. High Blood Press Cardiovasc Prev 2020;27:121-8.
5. Russo E, Viazzi F, Pontremoli R, et al. Uric acid and its relationship with eGFR and other correlates: the Uric Acid Right for heArt Health (URRAH) Project. J Nephrol. Forthcoming 2021.
6. Bonino B, Leoncini G, Russo E, Pontremoli R, Viazzi F. Uric acid in CKD: has the jury come to the verdict? J Nephrol 2020;33:715-24.
7. Johnson RJ, Bakris GL, Borghi C, et al. Hyperuricemia, acute and chronic kidney disease, hypertension, and cardiovascular disease: Report of a scientific workshop organized by the National Kidney Foundation. Am J Kidney Dis 2018;71:851-65.
8. Srivastava A, Kaze AD, McMullan CJ, Isakova T, Waikar SS. Uric acid and the risks of kidney failure and death in individuals with CKD. Am J Kidney Dis 2018;71:362-370.
9. Zhu P, Liu Y, Han L, Xu G, Ran JM. Serum uric acid is associated with incident chronic kidney disease in middle-aged populations: a meta-analysis of 15 cohort studies. PLoS One 2014;9:e100801.
10. Singh AK, Kari JA. Metabolic syndrome and chronic kidney disease. Curr Opin Nephrol Hypertens 2013;22:198-203.
11. Sharma K, Cook A, Smith M, Valancius C, Inscho EW. TGF-beta impairs renal autoregulation via generation of ROS. Am J Physiol Renal Physiol 2005;288:F1069-77.
12. Zager RA, Johnson A. Renal cortical cholesterol accumulation is an integral component of the systemic stress response. Kidney Int 2001;60:2299-310.
13. Park S, Chang YH, Cho YJ, et al. Cytokine-regulated expression of vascular cell adhesion molecule-1 in human glomerular endothelial cells. Transplant Proc 1998;30:2395-7.
14. Coleman DL, Ruef C. Interleukin-6: an autocrine regulator of mesangial cell growth. Kidney Int 1992;41:604-6.
15. Nakamura T, Miller D, Ruoslahti E, Border WA. Production of extracellular matrix by glomerular epithelial cells is regulated by transforming growth factor-beta 1. Kidney Int 1992;41:1213-21.
16. Khosla UM, Zharikov S, Finch JL, et al. Hyperuricemia induces endothelial dysfunction. Kidney Int 2005;67:1739-42.
17. Rao GN, Corson MA, Berk BC. Uric acid stimulates vascular smooth muscle cell proliferation by increasing platelet-derived growth factor A-chain expression. J Biol Chem 1991;266:8604-8.
18. Chao HH, Liu JC, Lin JW, Chen CH, Wu CH, Cheng TH. Uric acid stimulates endothelin-1 gene expression associated with NADPH oxidase in human aortic smooth muscle cells. Acta Pharmacol Sin 2008;29:1301-12.
19. Yu MA, Sánchez-Lozada LG, Johnson RJ, Kang DH. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J Hypertens 2010;28:1234-42.
20. Weiner DE, Tighiouart H, Elsayed EF, et al. Uric acid and incident kidney disease in the community. J Am Soc Nephrol 2008;19:1204-11.
21. Obermayr RP, Temml C, Gutjahr G, et al. Elevated uric acid increases the risk for kidney disease. J Am Soc Nephrol 2008;19:2407-13.
22. Braga TT, Forni MF, Correa-Costa M, et al. Soluble uric acid activates the NLRP3 inflammasome. Sci Rep 2017;7:39884.
23. Braga TT, Foresto-Neto O, Camara NOS. The role of uric acid in inflammasome-mediated kidney injury. Curr Opin Nephrol Hypertens 2020;29:423-431.
24. Alberts BM, Barber JS, Sacre SM, et al. Precipitation of soluble uric acid is necessary for in vitro activation of the NLRP3 inflammasome in primary human monocytes. J Rheumatol 2019;46:1141-50.
25. Välimäki E, Miettinen JJ, Lietzén N, Matikainen S, Nyman TA. Monosodium urate activates Src/Pyk2/PI3 kinase and cathepsin dependent unconventional protein secretion from human primary macrophages. Mol Cell Proteomics 2013;1:749-63.
26. Mulay SR, Anders HJ. Crystal nephropathies: mechanisms of crystal-induced kidney injury. Nat Rev Nephrol 2017;13:226-240.
27. Wen L, Yang H, Ma L, Fu P. The roles of NLRP3 inflammasome-mediated signaling pathways in hyperuricemic nephropathy. Mol Cell Biochem 2021; doi: 10.1007/s11010-020-03997-z.
28. Maejima I, Takahashi A, Omori H, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 2013;32:2336-47.
29. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002;10:417-26.
30. Chen J, Chen ZJ. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018;564:71-6.
31. Conforti-Andreoni C, Spreafico R, Qian HL, et al. Uric acid-driven Th17 differentiation requires inflammasome-derived IL-1 and IL-18. J Immunol 2011;187:5842-50.
32. Yen JH, Lin LC, Chen MC, et al. The metastatic tumor antigen 1-transglutaminase-2 pathway is involved in self-limitation of monosodium urate crystal-induced inflammation by upregulating TGF-β1. Arthritis Res Ther 2015;17:65.
33. Wang YJ, Chen YY, Hsiao CM, et al. Induction of autophagy by pterostilbene contributes to the prevention of renal fibrosis via attenuating NLRP3 inflammasome activation and epithelial-mesenchymal transition. Front Cell Dev Biol 2020;8:436.
34. Sader F, Denis JF, Laref H, Roy S. Epithelial to mesenchymal transition is mediated by both TGF-β canonical and non-canonical signaling during axolotl limb regeneration. Sci Rep 2019;9:1144.
35. Zavadil J, Böttinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005;24:5764-74.
36. Wang K, Hu L, Chen JK. RIP3-deficience attenuates potassium oxonate-induced hyperuricemia and kidney injury. Biomed Pharmacother 2018;101:617-26.
37. Grande MT, Sánchez-Laorden B, López-Blau C, et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med 2015;21:989-97.
38. Xiong W, Meng XF, Zhang C. Inflammasome activation in podocytes: a new mechanism of glomerular diseases. Inflamm Res 2020;69:731-43.
39. Al-Awad D, Al-Emadi N, Abu-Madi M, Al-Thani AA, Zughaier SM. The role of soluble uric acid in modulating autophagy flux and inflammasome activation during bacterial infection in macrophages. Biomedicines 2020;8:598.
40. Crișan TO, Cleophas MC, Oosting M, et al. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Ann Rheum Dis 2016;75:755-62.
41. Ghaemi-Oskouie F, Shi Y. The role of uric acid as an endogenous danger signal in immunity and inflammation. Curr Rheumatol Rep 2011;13:160-6.
42. Xiao J, Zhang XL, Fu C, et al. Soluble uric acid increases NALP3 inflammasome and interleukin-1β expression in human primary renal proximal tubule epithelial cells through the Toll-like receptor 4-mediated pathway. Int J Mol Med 2015;35:1347-54.
43. Xiao J, Fu C, Zhang X, et al. Soluble monosodium urate, but not its crystal, induces toll like receptor 4-dependent immune activation in renal mesangial cells. Mol Immunol 2015;66:310-8.
44. Gicquel T, Robert S, Loyer P, et al. IL-1β production is dependent on the activation of purinergic receptors and NLRP3 pathway in human macrophages. FASEB J 2015;29:4162-73.
45. Luo SF, Chin CY, Ho LJ, et al. Monosodium urate crystals induced ICAM-1 expression and cell-cell adhesion in renal mesangial cells: Implications for the pathogenesis of gouty nephropathy. J Microbiol Immunol Infect 2020;53:23-32.
46. Milanesi S, Verzola D, Cappadona F, et al. Uric acid and angiotensin II additively promote inflammation and oxidative stress in human proximal tubule cells by activation of toll-like receptor 4. J Cell Physiol 2019;234:10868-76.
47. Yang Y, Zhang DM, Liu JH, et al. Wuling San protects kidney dysfunction by inhibiting renal TLR4/MyD88 signaling and NLRP3 inflammasome activation in high fructose-induced hyperuricemic mice. J Ethnopharmacol 2015;169:49-59.
48. Ma WG, Wang J, Bu XW, et al. Effects of Polygonum cuspidatum on AMPK-FOXO3α signaling pathway in rat model of uric acid-induced renal damage. Chin J Integr Med 2019;25:182-9.
49. An MF, Wang MY, Shen C, et al. Isoorientin exerts a urate-lowering effect through inhibition of xanthine oxidase and regulation of the TLR4-NLRP3 inflammasome signaling pathway. J Nat Med 2021;75:129-41.
50. Han J, Shi G, Li W, Xie Y, Li F, Jiang D. Preventive effect of dioscin against monosodium urate-mediated gouty arthritis through inhibiting inflammasome NLRP3 and TLR4/NF-κB signaling pathway activation: an in vivo and in vitro study. J Nat Med 2021;75:37-47.
51. Ma JQ, Zhang YJ, Tian ZK, Liu CM. Bixin attenuates carbon tetrachloride induced oxidative stress, inflammation and fibrosis in kidney by regulating the Nrf2/TLR4/MyD88 and PPAR-γ/TGF-β1/Smad3 pathway. Int Immunopharmacol 2021;90:107117.
52. Hassanein EHM, Ali FEM, Kozman MR, Abd El-Ghafar OAM. Umbelliferone attenuates gentamicin-induced renal toxicity by suppression of TLR-4/NF-κB-p65/NLRP-3 and JAK1/STAT-3 signaling pathways. Environ Sci Pollut Res Int 2021;28:11558-71.
53. Shi M, Zeng X, Guo F, et al. Anti-inflammatory pyranochalcone derivative attenuates lps-induced acute kidney injury via inhibiting TLR4/NF-κB pathway. Molecules 2017;22:1683.
54. Poprac P, Jomova K, Simunkova M, Kollar V, Rhodes CJ, Valko M. Targeting free radicals in oxidative stress-related human diseases. Trends Pharmacol Sci 2017;38:592-607.
55. Zhou Y, You H, Zhang A, et al. Lipoxin A4 attenuates uric acid-activated, NADPH oxidase-dependent oxidative stress by interfering with translocation of p47phox in human umbilical vein endothelial cells. Exp Ther Med 2020;20:1682-92.
56. Momoki K, Kataoka H, Moriyama T, Mochizuki T, Nitta K. Hyperuricemia as a predictive marker for progression of nephrosclerosis: Clinical assessment of prognostic factors in biopsy-proven arterial/arteriolar nephrosclerosis. J Atheroscler Thromb 2017;24:630-42.
57. Convento MS, Pessoa E, Dalboni MA, Borges FT, Schor N. Pro-inflammatory and oxidative effects of noncrystalline uric acid in human mesangial cells: contribution to hyperuricemic glomerular damage. Urol Res 2011;39:21-7.
58. Corry DB, Eslami P, Yamamoto K, Nyby MD, Makino H, Tuck ML. Uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the vascular renin-angiotensin system. J Hypertens 2008;26:269-75.
59. Zhang JX, Zhang YP, Wu QN, Chen B. Uric acid induces oxidative stress via an activation of the renin-angiotensin system in 3T3-L1 adipocytes. Endocrine 2015;48:135-42.
60. Verzola D, Ratto E, Villaggio B, et al. Uric acid promotes apoptosis in human proximal tubule cells by oxidative stress and the activation of NADPH oxidase NOX 4. PLoS One 2014;9:e115210.
61. Roumeliotis S, Roumeliotis A, Dounousi E, Eleftheriadis T, Liakopoulos V. Dietary antioxidant supplements and uric acid in chronic kidney disease: A review. Nutrients 2019;11:1911.
62. Kanellis J, Kang DH. Uric acid as a mediator of endothelial dysfunction, inflammation, and vascular disease. Semin Nephrol 2005;25:39-42.
63. Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis 2006;47:51-9.
64. Goicoechea M, Garcia de Vinuesa S, Verdalles U, et al. Allopurinol and progression of CKD and cardiovascular events: long-term follow-up of a randomized clinical trial. Am J Kidney Dis 2015;65:543-9.
65. Kojima S, Matsui K, Hiramitsu S, et al. Febuxostat for Cerebral and CaRdiorenovascular Events PrEvEntion StuDy. Eur Heart J 2019;40:1778-86.
66. Su X, Xu B, Yan B, Qiao X, Wang L. Effects of uric acid-lowering therapy in patients with chronic kidney disease: A meta-analysis. PLoS One 2017;12:e0187550.
67. Kanellis J, Watanabe S, Li JH, et al. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension 2003;41:1287-93.
68. Kang DH, Park SK, Lee IK, Johnson RJ. Uric acid-induced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol 2005;16:3553-62.
69. Sánchez-Lozada LG, Tapia E, Santamaría J, et al. Mild hyperuricemia induces vasoconstriction and maintains glomerular hypertension in normal and remnant kidney rats. Kidney Int 2005;67:237-47.
71. Li Z, Sheng Y, Liu C, et al. Nox4 has a crucial role in uric acid induced oxidative stress and apoptosis in renal tubular cells. Mol Med Rep 2016;13:4343-8.
72. Yang Q, Wu FR, Wang JN, et al. Nox4 in renal diseases: An update. Free Radic Biol Med 2018;124:466-72.
73. Shirani M, Alizadeh S, Mahdavinia M, Dehghani MA. The ameliorative effect of quercetin on bisphenol A-induced toxicity in mitochondria isolated from rats. Environ Sci Pollut Res Int 2019;26:7688-96.
74. Sánchez-Lozada LG, Lanaspa MA, Cristóbal-García M, et al. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Exp Nephrol 2012;121:e71-8.
75. Lu H, Yao H, Zou R, Chen X, Xu H. Galangin Suppresses Renal Inflammation via the Inhibition of NF-κB, PI3K/AKT and NLRP3 in Uric Acid Treated NRK-52E Tubular Epithelial Cells. Biomed Res Int 2019;2019:3018357.
76. Yang L, Chang B, Guo Y, Wu X, Liu L. The role of oxidative stress-mediated apoptosis in the pathogenesis of uric acid nephropathy. Ren Fail 2019;41:616-22.
77. Uedono H, Tsuda A, Ishimura E, et al. Relationship between serum uric acid levels and intrarenal hemodynamic parameters. Kidney Blood Press Res 2015;40:315-22.
78. Canepa M, Viazzi F, Strait JB, Ameri P, Pontremoli R, et al. Longitudinal association between serum uric acid and arterial stiffness: Results from the Baltimore Longitudinal Study of aging. Hypertension 2017;69:228-35.
79. Kohagura K, Kochi M, Miyagi T, et al. An association between uric acid levels and renal arteriolopathy in chronic kidney disease: a biopsy-based study. Hypertens Res 2013;36:43-9.
80. Geraci G, Mulè G, Mogavero M, Geraci C, Nardi E, Cottone S. Association between uric acid and renal hemodynamics: Pathophysiological implications for renal damage in hypertensive patients. J Clin Hypertens (Greenwich) 2016;18:1007-14.
81. Aroor AR, Jia G, Habibi J, et al. Uric acid promotes vascular stiffness, maladaptive inflammatory responses and proteinuria in western diet fed mice. Metabolism 2017;74:32-40.
82. Zhang Y, He D, Zhang W, et al. ACE inhibitor benefit to kidney and cardiovascular outcomes for patients with non-dialysis chronic kidney disease stages 3-5: A network meta-analysis of randomised clinical trials. Drugs 2020;80:797-811.
83. Mikolajczyk TP, Szczepaniak P, Vidler F, Maffia P, Graham GJ, Guzik TJ. Role of inflammatory chemokines in hypertension. Pharmacol Ther 2020;223:107799.
84. Perlstein TS, Gumieniak O, Hopkins PN, et al. Uric acid and the state of the intrarenal renin-angiotensin system in humans. Kidney Int 2004;66:1465-70.
85. Mazzali M, Kanellis J, Han L, et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol Renal Physiol 2002;282:F991-7.
86. Rüster C, Wolf G. Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J Am Soc Nephrol 2011;22:1189-99.
87. Wang TN, Chen X, Li R, et al. SREBP-1 mediates angiotensin II-induced TGF-β1 upregulation and glomerular fibrosis. J Am Soc Nephrol 2015;26:1839-54.
88. Obama T, Eguchi S. MicroRNA as a novel component of the tissue renin angiotensin system. J Mol Cell Cardiol 2014;75:98-9.
89. Kanbay M, Sánchez-Lozada LG, Franco M, et al. Microvascular disease and its role in the brain and cardiovascular system: a potential role for uric acid as a cardiorenal toxin. Nephrol Dial Transplant 2011;26:430-7.
90. Aldivielso JM, Rodríguez-Puyol D, Pascual J, et al. Atherosclerosis in chronic kidney disease: more, less, or just different? Arterioscler Thromb Vasc Biol 2019;39:1938-66.
91. Myllymäki J, Honkanen T, Syrjänen J, et al. Uric acid correlates with the severity of histopathological parameters in IgA nephropathy. Nephrol Dial Transplant 2005;20:89-95.
92. Russo E, Drovandi S, Salvidio G, et al. Increased serum uric acid levels are associated to renal arteriolopathy and predict poor outcome in IgA nephropathy. Nutr Metab Cardiovasc Dis 2020;30:2343-50.
93. Zhang J, Diao B, Lin X, Xu J, Tang F. TLR2 and TLR4 mediate an activation of adipose tissue renin-angiotensin system induced by uric acid. Biochimie 2019;162:125-33.
94. Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001;38:1101-6.
95. Nakagawa T, Mazzali M, Kang DH, et al. Hyperuricemia causes glomerular hypertrophy in the rat. Am J Nephrol 2003;23:2-7.
96. South AM, Shaltout HA, Nixon PA, et al. Association of circulating uric acid and angiotensin-(1-7) in relation to higher blood pressure in adolescents and the influence of preterm birth. J Hum Hypertens 2020;34:818-25.
97. Hong Q, Wang L, Huang Z, et al. High concentrations of uric acid and angiotensin II act additively to produce endothelial injury. Mediators Inflamm 2020;2020:8387654.
98. Huang Z, Hong Q, Zhang X, et al. Aldose reductase mediates endothelial cell dysfunction induced by high uric acid concentrations. Cell Commun Signal 2017;15:3.
99. Hong Q, Yu S, Geng X, Duan L, Zheng W, et al. High concentrations of uric acid inhibit endothelial cell migration via miR-663 which regulates phosphatase and tensin homolog by targeting transforming growth factor-β1. Microcirculation 2015;22:306-14.
100. Yu S, Hong Q, Wang Y, et al. High concentrations of uric acid inhibit angiogenesis via regulation of the Krüppel-like factor 2-vascular endothelial growth factor-A axis by miR-92a. Circ J 2015;79:2487-98.
101. Mishima M, Hamada T, Maharani N, et al. Effects of uric acid on the NO production of HUVECs and its restoration by urate lowering agents. Drug Res (Stuttg) 2016;66:270-4.
102. Sugihara S, Hisatome I, Kuwabara M, et al. Depletion of uric acid due to SLC22A12 (URAT1) loss-of-function mutation causes endothelial dysfunction in hypouricemia. Circ J 2015;79:1125-32.
103. Li P, Zhang L, Zhang M, Zhou C, Lin N. Uric acid enhances PKC-dependent eNOS phosphorylation and mediates cellular ER stress: A mechanism for uric acid-induced endothelial dysfunction. Int J Mol Med 2016;37:989-97.
104. Spiga R, Marini MA, Mancuso E, et al. Uric acid is associated with inflammatory biomarkers and induces inflammation via activating the NF-κB Signaling Pathway in HepG2 Cells. Arterioscler Thromb Vasc Biol 2017;37:1241-9.
105. Zhen H, Gui F. The role of hyperuricemia on vascular endothelium dysfunction. Biomed Rep 2017;7:325-330.
106. Cai W, Duan XM, Liu Y, et al. Uric acid induces endothelial dysfunction by activating the HMGB1/RAGE signaling pathway. Biomed Res Int 2017;2017:4391920.
107. Oberbach A, Neuhaus J, Jehmlich N, et al. A global proteome approach in uric acid stimulated human aortic endothelial cells revealed regulation of multiple major cellular pathways. Int J Cardiol 2014;176:746-52.
108. Feng B, Zhang YQ, Mu J, et al. Uraemic serum induces dysfunction of vascular endothelial cells: role of ubiquitin-proteasome pathway. Exp Physiol 2011;96:801-15.
109. Xu J, Wang S, Wu Y, Song P, Zou MH. Tyrosine nitration of PA700 activates the 26S proteasome to induce endothelial dysfunction in mice with angiotensin II-induced hypertension. Hypertension 2009;54:625-32.
110. Doehner W, Schoene N, Rauchhaus M, et al. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo-controlled studies. Circulation 2002;105:2619-24.
111. Farquharson CA, Butler R, Hill A, Belch JJ, Struthers AD. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation 2002;106:221-6.
112. Ryu ES, Kim MJ, Shin HS, et al. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am J Physiol Renal Physiol 2013;304:F471-80.
113. Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol 2004;15:1-12.
114. Sheng L, Zhuang S. New insights into the role and mechanism of partial epithelial-mesenchymal transition in kidney fibrosis. Front Physiol 2020;11:569322.
115. Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 2008;19:2282-7.
116. Montorfano I, Becerra A, Cerro R, et al. Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-β1 and TGF-β2-dependent pathway. Lab Invest 2014;94:1068-82.
117. Pérez L, Muñoz-Durango N, Riedel CA, et al. Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev 2017;33:41-54.
118. Ko J, Kang HJ, Kim DA, et al. Uric acid induced the phenotype transition of vascular endothelial cells via induction of oxidative stress and glycocalyx shedding. FASEB J 2019;33:13334-45.
119. Reines BP, Ninham BW. Structure and function of the endothelial surface layer: unraveling the nanoarchitecture of biological surfaces. Q Rev Biophys 2019;52:e13.
120. Teeffelen JW, Brands J, Stroes ES, Vink H. Endothelial glycocalyx: sweet shield of blood vessels. Trends Cardiovasc Med 2007;17:101-5.
121. Noble MI, Drake-Holland AJ, Vink H. Hypothesis: arterial glycocalyx dysfunction is the first step in the atherothrombotic process. QJM 2008;101:513-8.
122. Su HY, Yang C, Liang D, Liu HF. Research advances in the mechanisms of hyperuricemia-induced renal injury. Biomed Res Int 2020;2020:5817348.
123. Shankar A, Klein BE, Nieto FJ, Klein R. Association between serum uric acid level and peripheral arterial disease. Atherosclerosis 2008;196:749-55.
124. Feig DI, Mazzali M, Kang DH, et al. Serum uric acid: a risk factor and a target for treatment? J Am Soc Nephrol 2006;17:S69-73.
125. Zoccali C, Maio R, Mallamaci F, Sesti G, Perticone F. Uric acid and endothelial dysfunction in essential hypertension. J Am Soc Nephrol 2006;17:1466-71.
126. Nakagawa T, Mazzali M, Kang DH, Sánchez-Lozada LG, Herrera-Acosta J, Johnson RJ. Uric acid--a uremic toxin? Blood Purif 2006;24:67-70.
127. Fu X, Niu N, Li G, et al. Blockage of macrophage migration inhibitory factor (MIF) suppressed uric acid-induced vascular inflammation, smooth muscle cell de-differentiation, and remodeling. Biochem Biophys Res Commun 2019;508:440-4.
128. Kang DH, Nakagawa T, Feng L, et al. A role for uric acid in the progression of renal disease. J Am Soc Nephrol 2002;13:2888-97.
129. Kırça M, Oğuz N, Çetin A, Uzuner F, Yeşilkaya A. Uric acid stimulates proliferative pathways in vascular smooth muscle cells through the activation of p38 MAPK, p44/42 MAPK and PDGFRβ. J Recept Signal Transduct Res 2017;37:167-73.
130. Schmeisser A, Marquetant R, Illmer T, et al. The expression of macrophage migration inhibitory factor 1alpha (MIF 1alpha) in human atherosclerotic plaques is induced by different proatherogenic stimuli and associated with plaque instability. Atherosclerosis 2005;178:83-94.
131. Lilly B. We have contact: endothelial cell-smooth muscle cell interactions. Physiology (Bethesda) 2014;29:234-41.
132. Marcelo KL, Goldie LC, Hirschi KK. Regulation of endothelial cell differentiation and specification. Circ Res 2013;112:1272-87.
133. Donovan J, Abraham D, Norman J. Platelet-derived growth factor signaling in mesenchymal cells. Front Biosci (Landmark Ed) 2013;18:106-19.
134. Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov 2012;11:790-811.
135. Baker AB, Ettenson DS, Jonas M, Nugent MA, Iozzo RV, Edelman ER. Endothelial cells provide feedback control for vascular remodeling through a mechanosensitive autocrine TGF-beta signaling pathway. Circ Res 2008;103:289-97.
136. Badri KR, Yue M, Carretero OA, et al. Blood pressure homeostasis is maintained by a P311-TGF-β axis. J Clin Invest 2013;123:4502-12.
137. Triggle CR, Samuel SM, Ravishankar S, Marei I, Arunachalam G, Ding H. The endothelium: influencing vascular smooth muscle in many ways. Can J Physiol Pharmacol 2012;90:713-38.
138. Boulos N, Helle F, Dussaule JC, et al. Notch3 is essential for regulation of the renal vascular tone. Hypertension 2011;57:1176-82.
139. Wu Z, Luo H, Thorin E, et al. Possible role of Efnb1 protein, a ligand of Eph receptor tyrosine kinases, in modulating blood pressure. J Biol Chem 2012;287:15557-69.
140. Viazzi F, Rebora P, Giussani M, et al. Increased serum uric acid levels blunt the antihypertensive efficacy of lifestyle modifications in children at cardiovascular risk. Hypertension 2016;67:934-40.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Russo E, Verzola D, Cappadona F, Leoncini G, Garibotto G, Pontremoli R, Viazzi F. The role of uric acid in renal damage - a history of inflammatory pathways and vascular remodeling. Vessel Plus 2021;5:15. http://dx.doi.org/10.20517/2574-1209.2021.11
AMA Style
Russo E, Verzola D, Cappadona F, Leoncini G, Garibotto G, Pontremoli R, Viazzi F. The role of uric acid in renal damage - a history of inflammatory pathways and vascular remodeling. Vessel Plus. 2021; 5: 15. http://dx.doi.org/10.20517/2574-1209.2021.11
Chicago/Turabian Style
Russo, Elisa, Daniela Verzola, Francesca Cappadona, Giovanna Leoncini, Giacomo Garibotto, Roberto Pontremoli, Francesca Viazzi. 2021. "The role of uric acid in renal damage - a history of inflammatory pathways and vascular remodeling" Vessel Plus. 5: 15. http://dx.doi.org/10.20517/2574-1209.2021.11
ACS Style
Russo, E.; Verzola D.; Cappadona F.; Leoncini G.; Garibotto G.; Pontremoli R.; Viazzi F. The role of uric acid in renal damage - a history of inflammatory pathways and vascular remodeling. Vessel Plus. 2021, 5, 15. http://dx.doi.org/10.20517/2574-1209.2021.11
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 16 clicks
Cite This Article 16 clicks

 Like This Article 23
likes
Like This Article 23
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.