
A failure of forward translation? The case of neuroprotection
 0
0Abstract
More than half a century of research focused on ischemic brain injury mechanisms has failed to yield a widely accepted neuroprotective drug for the treatment of acute ischemic stroke (AIS). The absence of a therapeutic intervention targeted at neuroprotective mechanisms raises questions about the relevance of preclinical models in human stroke. Indeed, this failure of forward translation (traditional bench-to-bedside research) to bring candidate drugs into clinical use suggests that alternative or complementary approaches are needed. Here, we discuss the potential of reverse translational research - exploring a bedside-to-bench approach - utilizing big data genomics to discover novel AIS therapeutic targets. This approach might provide insights into new and old drug targets.
Keywords
Stroke and neuroprotection
Worldwide, stroke is the second leading cause of death and the most common cause of adult disability[1,2]. Recent advances in reperfusion therapies (thrombolysis and thrombectomy) have dramatically improved acute stroke care (as discussed in other articles in this issue), impacting long term outcomes. Despite the positive impact of reperfusion-based therapies, acute ischemic stroke (AIS) patients continue to suffer substantial disability. Among patients treated with mechanical thrombectomy, 50% are functionally dependent and 75% are disabled at 90 days[3-5]. Thus, there is critical need to continue to develop novel therapies that can either enhance reperfusion or improve outcomes regardless of reperfusion. Among these additional targets is neuroprotection - the strategy of interfering with the ischemic cascades by blocking cellular and molecular pathways leading to neuronal cell death. Targets of neuroprotection include excitotoxicity - the pathological excitation of neurons due to the massive release of the excitatory neurotransmitter glutamate-cellular influx of calcium, generation of free radicals, and inflammatory cascades[6,7]. However, failure of translation from experimental models to successful human trials has virtually halted the drug development pipeline for neuroprotection. As a result, investment by pharmaceutical companies in this market has been limited. However, in this new era of reperfusion therapy, neuroprotection is being actively reconsidered, with the premise that earlier trials failed because neuroprotectants were unable to reach ischemic tissue in adequate concentrations[8].
Failure of forward translation
Despite five decades of translational research on ischemic brain injury mechanisms, there are no widely accepted neuroprotective drugs for the treatment of AIS. It has been estimated that over a thousand drug targets have been identified from cellular or animal models[9]. Of these, less than 100 have been tested in human clinical stroke trials[9], and virtually all trials have been negative. These poor odds identify the translation from preclinical studies to clinical trials as a bottleneck in identifying drug targets relevant to human disease [Figure 1A].
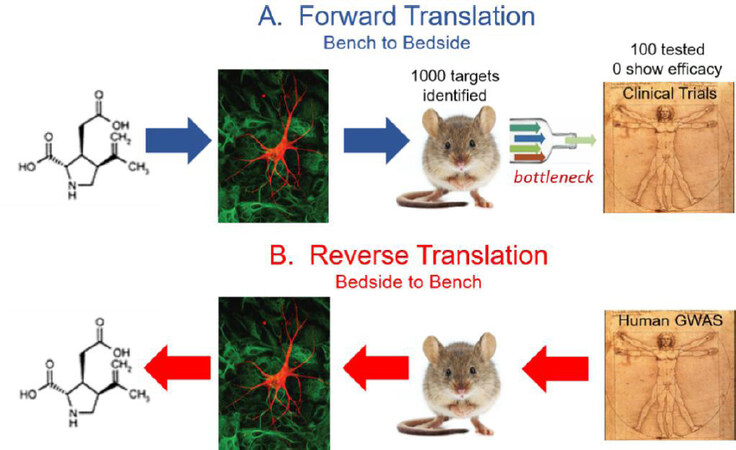
Figure 1. The failure of forward translation-neuroprotection. Five decades of translation research on acute ischemic brain injury mechanisms have failed to produce a widely accepted neuroprotective drug for the treatment of acute ischemic stroke. A: Over 1,000 drug targets have been identified, yet only 100 have been tested in clinical trials-none have proven efficacious. Thus, translation from preclinical animal models to clinical trials appears to be a bottleneck in forward translation. Alternative approaches are needed to confirm the relevance of drug targets in human stroke, or to discover novel drug targets. B: Here we explore the feasibility and effectiveness of reverse translation approaches-in particular genome-wide association studies-in their ability to confirm old drug targets or reveal novel targets.
These trial failures led to a period of deep introspection in the field, with many questioning the validity of preclinical animal models for discovering novel drug targets for translation to human clinical trials[10-12]. The discussion also stimulated a re-examination of the rigor of early stroke trials, which often did not confirm target engagement or adhere to relevant therapeutic time windows[13-15]. To enhance the translational potential of new experimental therapies, the Stroke Therapy Academic Industry Roundtable (STAIR) published guidelines in 1999 to develop rigorous criteria for preclinical studies in animal models. Among the recommendations were pretrial sample size calculations, randomization, blinded allocation, and endpoint assessments, the inclusion of aged animals with comorbid conditions[16]. However, preclinical studies that adhered to STAIR criteria still failed in subsequent clinical trials[17,18]. Despite updated STAIR criteria[19], we have yet to have a positive clinical trial for neuroprotection.
It is clear that the traditional approach of forward translation - starting at the bench with the identification of potential drug targets and translating to clinical trials - is costly and inefficient. Are there alternative or complementary approaches towards validating drug targets in humans?
Reverse translation - GWAS
Over the last decade, genome-wide-association studies (GWAS) have identified thousands of genetic variants that are associated with human traits and diseases. GWAS takes advantage of natural variation in the human genome to identify genetic markers that associate with specific traits or diseases. Unlike candidate gene approaches, which examine the association between a given trait and select candidate genes, GWAS examines associations between the trait and genetic markers across the entire genome, creating a large-scale unbiased approach. Over 3,000 human GWAS have investigated more than 1,800 diseases and traits yielding thousands of genetic associations[20]. Examples of diseases that have led to the discovery of genes and pathways involved in pathogenesis include age-related macular degeneration[21], inflammatory bowel disease[22], cardiovascular disease[23], obesity[24], schizophrenia[25], and Alzheimer disease[26]. While these genetic associations are often weak, accounting for only a small amount of the risk for the disease, they provide important insights into genes and pathways that are involved in disease pathogenesis. Moreover, GWAS studies examining genetic overlap between disparate diseases may also shed light on common pathogenic relationships based on shared genetic mechanisms[27]. Many of these genetic associations have confirmed old drug targets or identified potentially novel drug targets[28,29][Figure 1B].
Indeed, big pharma has realized the importance of human genetics in identifying therapeutic targets for disease. Recent retrospective reviews of the drug pipeline at several large pharmaceutical companies have revealed that if a drug target is independently confirmed using human genetics, the drug is twice as likely to attain FDA-approval[30,31].
Genetics of early neurological instability after ischemic stroke
Towards that end, we recently completed the genetics of early neurological instability after ischemic stroke (GENISIS) study - a GWAS of 5,876 AIS patients, examining genetic associations with early neurological change within the first 24 h after stroke onset (ΔNIHSS = NIHSS6h-NIHSS24h). This dynamic metric of neurological change captures both early deterioration (negative ΔNIHSS) and early improvement (positive ΔNIHSS) following AIS. ΔNIHSS falls into a normal distribution and segregates specific AIS mechanisms along the spectrum of quantitative scores[32]. For example, extreme negative ΔNIHSS (deterioration) is associated with hemorrhagic transformation; while extreme positive ΔNIHSS (improvement) is often associated with recanalization (in a sub-cohort of patients with large vessel occlusion)[32]. We hypothesized that using ΔNIHSS as a quantitative trait with GWAS would reveal genetic variants, genes, or pathways related to early ischemic brain injury mechanisms, and provide insight into potential drug targets.
AIS patients were prospectively enrolled from more than 20 sites from seven countries throughout the world, including Asia, Europe, North America, and South America. The varied cohorts from multi-ethnic populations is advantageous for genomic studies because of the inclusion of individuals with wide genetic diversity[33]. However, multi-ethnic cohorts also pose challenges to genetic analyses due to the substantial heterogeneity in genetic architecture across diverse populations[34]. To overcome this obstacle, we performed a multi-ancestry Bayesian meta-analysis (MANTRA) to account for differences in population structures[35].
The MANTRA GWAS revealed seven genome-wide significant loci associated with ΔNIHSS: three loci on chromosome 2 (2p25.1, 2q31.2, and 2q33.3), and one locus on chromosome 4 (4q34.3), 5 (5q33.2), 6 (6q26), and 7 (7p21.1)[36]. One of the top loci, located on chromosome 2 (2q33.3), fell within the gene, ADAM23. Expression quantitative trait loci (eQTL) analysis, using a variety of eQTL databases (GTEx portal and Braineac), confirmed that this genetic locus influenced expression of ADAM23 in different tissue including brain. Mendelian randomization, a method used to examine causal effects of genetic variants on quantitative traits, revealed that a SNP associated with ADAM23 expression was also associated with ΔNIHSS. Finally, single-nuclei RNA-seq data from parietal lobes of 68 post-mortem brains (from subjects free of neurological disorders)[37] revealed that ADAM23 was largely expressed in neurons, and predominantly in excitatory neurons[36]. A second strong genome-wide association was found in chromosome 5 (5q33.2), falling within the gene, GRIA1, which encodes the AMPA-receptor subtype 1. Gene-based analysis using FUMA revealed that GRIA1 was the gene most likely driving the association to the 5q33.2 locus [Table 1]. Moreover, single-nuclei RNA-seq from the human post-mortem brains, demonstrated that GRIA1 was exclusively expressed in neurons[36].
Lead genome-wide associated SNPs based on MANTRA
| Chromosome | SNP | Minor allele frequency | Log Bayes factor* | Directionality by cohort† | Candidate genes |
|---|---|---|---|---|---|
| 2 | rs13403787 | 0.1578 | 5.57328 | +?++ | RNF144A |
| rs72958644 | 0.03562 | 6.34263 | +?+? | DFNB59 | |
| rs58763243 | 0.92954 | 6.50673 | ++-+ | ADAM23 | |
| 4 | rs12641856 | 0.07425 | 5.49467 | -?++ | MGC45800 |
| 5 | rs114248865 | 0.0536 | 5.28917 | +?+? | GRIA1 |
| 6 | rs6930598 | 0.09936 | 5.30039 | ---+ | PARK2 |
| 7 | rs10807797 | 0.5794 | 5.69768 | ++++ | TWISTNB ABCB5 |
We tentatively mapped four of the remaining five loci. Associated SNPs at the 2q31.2 and 4q34.3 loci were determined to be eQTLs for DFNB59 and MGC45800, respectively, implicating the involvement of these genes in early neurological change. No eQTLs were identified for 6q26; however, the locus fell within the boundaries of PARK2-a gene implicated in Parkinson’s disease and mitochondrial function[38]. Finally, 7p21.1 was associated with several eQTLs including TWISTNB and ABCB5[Table 1][36].
ADAM23 & GRIA1 - implications for excitotoxicity
ADAM23 belongs to a family of transmembrane proteins (a disintegrin and metalloproteinase)[39]. ADAM23 lacks protease activity but plays a role in cell-cell interactions. It is expressed on synaptic membranes in neurons and appears to bridge pre- and post-synaptic terminals. Pre-synaptic ADAM23 binds to the extracellular protein LGI1 (leucine rich glioma 1), which in turn binds to postsynaptic ADAM22 (another closely related ADAM family member)[40,41]. This bridge brings the presynaptic vesicular release machinery in close apposition to the postsynaptic receptor scaffold (PSD-95), thereby regulating trans-synaptic excitability[42]. ADAM23 also clusters together voltage-gate potassium channels at the presynaptic membrane [Figure 2], thereby enhancing membrane repolarization. Disruption of ADAM23’s interaction with LGI1 causes excess glutamate release, likely as a result of dispersion of potassium channels important for repolarization[43]. Thus ADAM23 function at glutamate synapses might be considered to restrain glutamate release. Indeed, several diseases related to neuronal excitability are associated with the disruption of LGI1 binding to ADAM22/23: (1) a genetic mutation in LGI1 leads to autosomal dominant partial epilepsy with auditory features in humans[44]; (2) auto-antibodies directed against LGI1 lead to limbic encephalitis and seizures [Figure 2][42]; and (3) adam23 was found to be a common risk gene for canine idiopathic epilepsy[45].
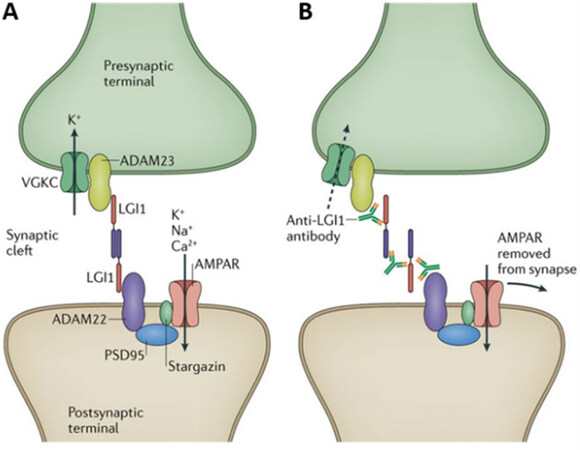
Figure 2. ADAM23 and GRIA1 (AMPA-R subtype 1) are part of a trans-synaptic protein complex that regulates synaptic excitability (A). Disruption of the protein complex with autoantibodies (B) or genetic mutation, results in hyper-excitability, seizures, and encephalopathy. Reproduced with permission from Springer/Nature Review Neurology[42].
Remarkably, one of our other genome-wide associated genes, GRIA1, appears to play a similar role regulating synaptic excitability. Indeed, GRIA1, which encodes for the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor subunit 1 (AMPAR1), is a known binding partner to ADAM23 via ADAM22 and PSD95 [Figure 2][46]. It has long been known that AMPA receptors, along with other glutamate receptors, are mediators of excitotoxic neuronal death, hypothesized to play an important role in ischemic brain injury[7,47]. As discussed above, failure of numerous older clinical trials examining the efficacy of anti-excitotoxic drugs had cast doubt on the relevance of excitotoxicity in human AIS[48,49]. However, this newly discovered association between the genes ADAM23 and GRIA1, and ΔNIHSS provides the first genetic evidence that excitotoxicity may contribute to ischemic brain injury in humans.
Future of reverse translation in acute ischemic stroke
The plausible roles that ADAM23 and GRIA1 play in acute brain ischemia mechanisms provide proof of principle that GWAS using ΔNIHSS as a quantitative phenotype can identify mechanisms and potential drug targets to mitigate neurological deterioration or enhance early improvement after stroke. In addition to the two genes discussed above, five other genetic loci were identified, whose functional genes remain to be identified. In the GENESIS study, common variants throughout the genome accounted for 8.7% of the variance of ΔNIHSS (SE 0.043; P = 0.001). The seven genetic loci discovered in this GWAS account for only 2.1% of this variance[36]. Therefore, many additional loci associated with ΔNIHSS remain to be discovered, requiring greater statistical power, provided by larger sample sizes and/or more biologically homogenous stroke cohorts.
In this age of thrombolysis and thrombectomy, AIS patients are well phenotyped as standard of care practice with both clinical and imaging assessments. Stroke patients with large vessel occlusion are particularly well-phenotyped, with vessel and perfusion imaging, providing time-dependent structural and physiological information about ongoing brain ischemia. Thus, there is great potential for additional quantitative phenotypes, including penumbral viability, collateral flow indices, edema formation, hemorrhagic transformation, and recanalization-dependent outcomes. Each phenotype promises to reveal distinct and overlapping genetic architectures that may uncover known and novel mechanisms involved in AIS.
Beyond genomics-multi-omics
Three decades of genome research and rigorous debates since the completion of the Human Genome Project have taught us that the causes of late-onset common diseases, even in high-risk populations with known major risk factors, are complex. To address this complex problem, one can more deeply characterize endophenotypes by tackling the central dogma of molecular biology: genes generating mRNA (transcriptomics), the translation of mRNA to proteins (proteomics), and the production of metabolites after post-translational modifications (metabolomics). Because these traits lie closer to the actions of the causal genes than to clinical outcomes, the relations between the gene(s) and these quantitative intermediate risk and protective factors will be much stronger than the relations between genetic factors and disease outcomes. By leveraging novel multi-omics approaches[50], links between genetic loci, specific mRNAs, proteins, and/or metabolites, will help identify more complete networks and pathways implicated in ischemic brain injury [Figure 3]. Identification of these pathways will be critical for the discovery of novel drug targets.
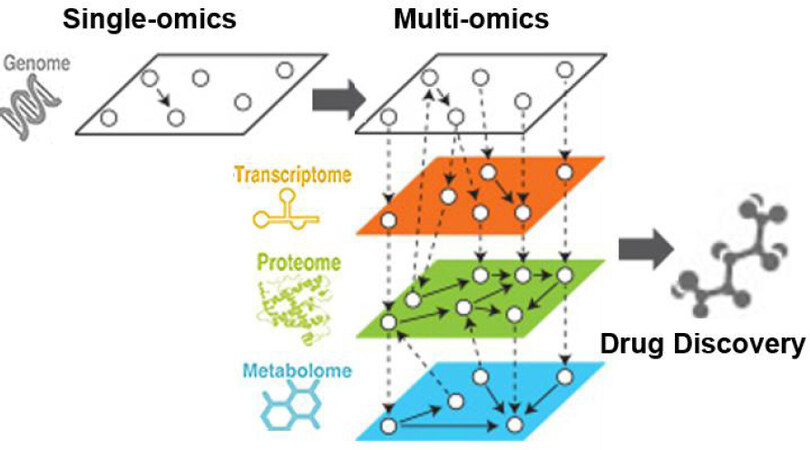
Figure 3. Beyond genomics to multi-omics. While GWAS has proven invaluable for linking genetic variants to disease phenotypes, a deeper understanding of underlying mechanisms may require the multiple layers of multi-omics. By linking pathways across genomes, transcriptomes, proteomes, and metabolomes, a more complex and complete set of pathways emerge, increasing power for discovery. GWAS: Genome-wide-association studies.
Conclusion and future directions
In summary, reverse translational approaches applied to AIS phenotypes promise to provide a rich adjunct to traditional forward translational approaches. The GENISIS study has demonstrated proof of principle that GWAS can be used with acute stroke phenotypes to discover mechanism involved in acute ischemic brain injury. This approach will be useful not only to confirm human relevance of drug targets discovered using forward translation but will also be useful for discovering novel mechanisms and drug targets. Future studies leveraging multi-omics promises to increase the dimensionality of pathogenic pathways linked to AIS, revealing even more novel mechanisms. Moreover, larger studies using more homogeneous subpopulations of stroke patients will be needed to increase power to detect many more genetic and mutli-omic associations to provide a more complete understanding of AIS pathogenesis. Thus, stroke translational research will benefit from fluidity in both forward and reverse directions.
Declarations
Authors’ contributionsConcepts and writing of this review article: Lee JM, Rosand J, Cruchaga C
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by NIH R01 NS085419, research support from Biogen.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol ;18:459-480.
2. Johnson W, Onuma O, Owolabi M, Sachdev S. Stroke: a global response is needed. Bull World Health Organ 2016;94:634-634A.
3. Albers GW, Marks MP, Kemp S, et al; DEFUSE 3 Investigators. Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging. N Engl J Med 2018;378:708-18.
4. Goyal M, Menon BK, van Zwam WH, et al. Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet 2016;387:1723-31.
5. Jovin TG, Chamorro A, Cobo E, et al; REVASCAT Trial Investigators. Thrombectomy within 8 hours after symptom onset in ischemic stroke. N Engl J Med 2015;372:2296-306.
6. Lee JM, Grabb MC, Zipfel GJ, Choi DW. Brain tissue responses to ischemia. J Clin Invest 2000;106:723-31.
7. Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature 1999;399:A7-14.
9. O’Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol 2006;59:467-77.
10. Degraba TJ, Pettigrew LC. Why do neuroprotective drugs work in animals but not humans? Neurologic Clinics 2000;18:475-93.
11. Philip M, Benatar M, Fisher M, Savitz SI. Methodological quality of animal studies of neuroprotective agents currently in phase II/III acute ischemic stroke trials. Stroke 2009;40:577-81.
12. Schmidt-Pogoda A, Bonberg N, Koecke MHM, et al. Why most acute stroke studies are positive in animals but not in patients: a systematic comparison of preclinical, early phase, and phase 3 clinical trials of neuroprotective agents. Ann Neurol 2020;87:40-51.
13. Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology 2008;55:363-89.
14. Grupke S, Hall J, Dobbs M, Bix GJ, Fraser JF. Understanding history, and not repeating it. Neuroprotection for acute ischemic stroke: from review to preview. Clin Neurol Neurosurg 2015;129:1-9.
15. Herson PS, Traystman RJ. Animal models of stroke: translational potential at present and in 2050. Future Neurol 2014;9:541-51.
16. Stroke Therapy Academic Industry Roundtable (STAIR). Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 1999;30:2752-8.
17. Feuerstein GZ, Zaleska MM, Krams M, et al. Missing steps in the STAIR case: a Translational Medicine perspective on the development of NXY-059 for treatment of acute ischemic stroke. J Cereb Blood Flow Metab 2008;28:217-9.
18. Savitz SI, Fisher M. Future of neuroprotection for acute stroke: in the aftermath of the SAINT trials. Ann Neurol 2007;61:396-402.
19. Fisher M, Feuerstein G, Howells DW, et al; STAIR Group. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 2009;40:2244-50.
20. Visscher PM, Wray NR, Zhang Q, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet 2017;101:5-22.
21. DeAngelis MM, Owen LA, Morrison MA, et al. Genetics of age-related macular degeneration (AMD). Hum Mol Genet 2017;26:R45-50.
22. Furey TS, Sethupathy P, Sheikh SZ. Redefining the IBDs using genome-scale molecular phenotyping. Nat Rev Gastroenterol Hepatol 2019;16:296-311.
23. Zheng H, Zeng Z, Wen H, et al. Application of genome-wide association studies in coronary artery disease. Curr Pharm Des 2019;25:4274-86.
24. Rohde K, Keller M, la Cour Poulsen L, Blüher M, Kovacs P, Böttcher Y. Genetics and epigenetics in obesity. Metabolism 2019;92:37-50.
25. Horwitz T, Lam K, Chen Y, Xia Y, Liu C. A decade in psychiatric GWAS research. Mol Psychiatry 2019;24:378-89.
26. Andrews SJ, Fulton-howard B, Goate A. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurology 2020;19:326-35.
27. Chen Q, Zhou XJ, Sun F. Finding genetic overlaps among diseases based on ranked gene lists. J Comput Biol 2015;22:111-23.
28. Jhamb D, Magid-Slav M, Hurle MR, Agarwal P. Pathway analysis of GWAS loci identifies novel drug targets and repurposing opportunities. Drug Discov Today 2019;24:1232-6.
29. Parvathaneni V, Kulkarni NS, Muth A, Gupta V. Drug repurposing: a promising tool to accelerate the drug discovery process. Drug Discov Today 2019;24:2076-85.
30. Cook D, Brown D, Alexander R, et al. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat Rev Drug Discov 2014;13:419-31.
31. Nelson MR, Tipney H, Painter JL, et al. The support of human genetic evidence for approved drug indications. Nat Genet 2015;47:856-60.
32. Heitsch L, Ibanez L, Carrera C, et al. Early neurological change after ischemic stroke is associated with 90-day outcome. Stroke ;52:132-41.
33. Zaitlen N, Paşaniuc B, Gur T, Ziv E, Halperin E. Leveraging genetic variability across populations for the identification of causal variants. Am J Hum Genet 2010;86:23-33.
34. Cooper RS, Tayo B, Zhu X. Genome-wide association studies: implications for multiethnic samples. Hum Mol Genet 2008;17:R151-5.
35. Morris AP. Transethnic meta-analysis of genomewide association studies. Genet Epidemiol 2011;35:809-22.
36. Ibanez L, Heitsch L, Carrera C, et al. Multi-ancestry genetic study in 5,876 patients identifies an association between excitotoxic genes and early outcomes after acute ischemic stroke. medRxiv 2020:2020.
37. Del-Aguila JL, Li Z, Dube U, et al. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res Ther 2019;11:71.
38. Ge P, Dawson VL, Dawson TM. PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson’s disease. Mol Neurodegener 2020;15:20.
39. Zhong S, Khalil RA. A Disintegrin and Metalloproteinase (ADAM) and ADAM with thrombospondin motifs (ADAMTS) family in vascular biology and disease. Biochem Pharmacol 2019;164:188-204.
40. Hivert B, Marien L, Agbam KN, Faivre-Sarrailh C. ADAM22 and ADAM23 modulate the targeting of the Kv1 channel-associated protein LGI1 to the axon initial segment. J Cell Sci 2019;132:jcs219774.
41. Lancaster E, Burnor E, Zhang J, Lancaster E. ADAM23 is a negative regulator of Kv1.1/Kv1.4 potassium currents. Neurosci Lett 2019;704:159-63.
42. van Sonderen A, Petit-Pedrol M, Dalmau J, Titulaer MJ. The value of LGI1, Caspr2 and voltage-gated potassium channel antibodies in encephalitis. Nat Rev Neurol 2017;13:290-301.
43. Petit-Pedrol M, Sell J, Planagumà J, et al. LGI1 antibodies alter Kv1.1 and AMPA receptors changing synaptic excitability, plasticity and memory. Brain 2018;141:3144-59.
44. Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science 2006;313:1792-5.
45. Koskinen LL, Seppälä EH, Weissl J, et al. ADAM23 is a common risk gene for canine idiopathic epilepsy. BMC Genet 2017;18:8.
46. Lovero KL, Fukata Y, Granger AJ, Fukata M, Nicoll RA. The LGI1-ADAM22 protein complex directs synapse maturation through regulation of PSD-95 function. Proc Natl Acad Sci U S A 2015;112:E4129-37.
47. Palmer CL, Cotton L, Henley JM. The molecular pharmacology and cell biology of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Pharmacol Rev 2005;57:253-77.
48. Chamorro Á, Dirnagl U, Urra X, Planas AM. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. The Lancet Neurology 2016;15:869-81.
49. Neuhaus AA, Couch Y, Hadley G, Buchan AM. Neuroprotection in stroke: the importance of collaboration and reproducibility. Brain 2017;140:2079-92.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lee JM, Rosand J, Cruchaga C. A failure of forward translation? The case of neuroprotection. Vessel Plus 2021;5:8. http://dx.doi.org/10.20517/2574-1209.2020.72
AMA Style
Lee JM, Rosand J, Cruchaga C. A failure of forward translation? The case of neuroprotection. Vessel Plus. 2021; 5: 8. http://dx.doi.org/10.20517/2574-1209.2020.72
Chicago/Turabian Style
Lee, Jin-Moo, Jonathan Rosand, Carlos Cruchaga. 2021. "A failure of forward translation? The case of neuroprotection" Vessel Plus. 5: 8. http://dx.doi.org/10.20517/2574-1209.2020.72
ACS Style
Lee, J.M.; Rosand J.; Cruchaga C. A failure of forward translation? The case of neuroprotection. Vessel Plus. 2021, 5, 8. http://dx.doi.org/10.20517/2574-1209.2020.72
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 11 clicks
Cite This Article 11 clicks

 Like This Article 28
likes
Like This Article 28
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.