
Inhibition of cyclooxygenase activity by diclofenac attenuates varicose remodeling of mouse veins
 0
0Abstract
Aim: The development of varicose veins is driven by risk factors that support the progression of venous hypertension, specifically, by chronically augmenting the circumferential tension of the venous wall. We have previously verified the relevance of this biomechanical stimulus for the activation of venous cells and the structural remodeling of the vein wall. Recent transcriptome analyses revealed an increase in the expression of the gene encoding prostaglandin-endoperoxide synthase 2 [cyclooxygenase 2 (COX-2)] in biomechanically stressed human vein endothelial cells. This observation prompted us to investigate the functional relevance of COX activity for the onset of pressure-induced venous remodeling.
Methods: For the in vitro experiments, isolated mouse veins were exposed to elevated intraluminal pressure levels to study the markers of cellular activation. For the in vivo experiments, pressure-dependent varicose remodeling of veins was induced by ligation of an efferent vein in the mouse auricle. Diclofenac was applied to inhibit the activity of COX.
Results: Short-term exposure to elevated pressure levels stimulated the abundance of activated matrix-metalloproteinase-2 (MMP-2) and mitogen activated protein kinase, ERK1/2, in isolated mouse veins, which was inhibited upon treatment with diclofenac. Transdermal application of diclofenac-containing phospholipid-micelles attenuated the corkscrew-like enlargement of veins and decreased the abundance of COX-2 and MMP-2 as well as cell proliferation in the venous wall.
Conclusion: The cyclooxygenase inhibitor, diclofenac, interferes with stress-induced activation of venous cells and attenuates venous remodeling in vivo. Additional research is warranted to investigate whether nonsteroidal anti-inflammatory drugs interfere with the processes promoting the onset of varicose vein development and biomechanical activation of venous cells.
Keywords
Introduction
Varicose veins frequently develop in the veins of lower extremities including the superficial, deep or perforating veins[1], reflecting the relevance of pressure gradients such as biomechanical stimuli in this pathophysiological process. As such, prolonged standing and tight undergarments are considered as primary risk factors of the disease[2,3], leading to elevated venous filling pressure and consequently, increases in biomechanical stress to which the venous wall is exposed. Likewise, the development of pelvic venous insufficiency and varicosities in the context of pelvic congestion syndrome[4] may also be promoted by an increase in venous pressure due to the partial occlusion of pelvic veins. In the long run, any congestion of blood flow in large veins may cause chronic elevation of blood pressure in the connected venous network and elevate venous wall tension. This biomechanical force is sufficient to promote the detrimental and structural remodeling of the venous wall architecture in mouse models[5,6], and is thought to drive comparable processes in humans[7]. Morphologically, this results in the development of thin-walled varicose veins, further characterized by their corkscrew-like and tortuous appearance, enlarged diameter, altered extracellular matrix composition and a change in the activity of endothelial and smooth muscle cells[8]. At the molecular level, loss of the regular collagen-elastin network, changes in the collagen type I to III-ratio[9], and an elevated activity of matrix-metalloproteinases (MMPs) such as the gelatinases MMP-2 and MMP-9[9-11] are reported for varicose veins.
Recently, MMP activity and collagen content have been linked to the level of prostaglandin E2 (PGE2), a cyclooxygenase (COX)-derived prostanoid, in human varicose veins[12]. Moreover, elevated PGE2 levels[13] as well as increased expression of the prostaglandin-endoperoxide synthase gene (PTGS1/2; encoded by COX-1/2)[14] have been detected previously in the development of varicosities. In this context, biomechanical wall stress could serve as a determinant of PTGS2 expression, which is up-regulated in biomechanically stressed venous endothelial cells[15], and has been associated with the remodeling of the extracellular matrix and vascular stiffness of arteries in hypertensive rats[16]. Although there is evidence that demonstrates a general association between COX activity and varicose vein development, their functional relevance for the onset of pressure-induced venous remodeling has not been investigated so far.
Herein, we hypothesized that the activity of COX-1/2 promotes remodeling of the venous wall via hypertensive venous pressure levels and chronically elevated venous wall stress. Consequently, this study aimed to investigate the effect of the COX-1/2 inhibitor, diclofenac, on pressure-induced responses of veins and venous remodeling by employing in vitro and in vivo experimental mouse models.
Methods
HUVEC and huSMC culture
The procedure for isolating human umbilical vein endothelial cells (HUVECs) was carried out with the approval from the Local Ethical Committee (document number 336/2005, Heidelberg, Germany) and conformed to the principles outlined in the Declaration of Helsinki (1997). Isolation of HUVECs from the umbilical cords of newborns was carried out upon parental consent. HUVECs were cultured in EASY medium (PeloBiotech, Germany) supplemented with endothelial cell growth supplement and 5% fetal calf serum (FCS). For proliferation analyses, HUVECs were cultured in EASY medium supplemented with 15% FCS. Human umbilical smooth muscle cells (huSMCs) were purchased from Provitro AG (Germany) and cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 15% FCS. For proliferation analyses, huSMCs were cultured in DMEM supplemented with 5% FCS.
Stimulation of HUVEC by biomechanical stretch
Cells were cultured on BioFlex® Collagen type I 6-well plates (Flexcell, North Carolina, USA) pre-coated with Geltrex® (basement membrane surrogate, 1:10 in cell media; ThermoFisher Scientific, Massachusetts, USA) for 1 h at 37 °C. One day prior to the stretch, the endothelial cell supplement content of the media was reduced to half and diluted in M199 media supplemented with 12.5% FCS, 25 U/ml penicillin, 25 μg/ml streptomycin and 0.125 μg/ml Fungizone® antimycotic. Cyclic stretch was applied using a microprocessor controlled vacuum pump (FX-5000 FlexerCell® strain unit, Flexcell®, North Carolina, USA) with 15% cyclic elastomer elongation at a frequency of 0.5 Hz. Cyclic elongation, as opposed to static, is needed to prevent the cells from evading the biomechanical stimulus through rearranging their focal contacts. Cells were stretched at 85%-95% confluency.
DNA microarray
HUVECs from three different donors were exposed to biomechanical stretch for 6 h or cultured under static conditions (control). RNA was isolated and processed for DNA microarray analysis according to the manufacturers’ instructions: Gene expression profiling was performed using the GeneChip® Human Genome Array from Affymetrix (Thermo Fisher Scientific Inc). After RNA isolation, RNA was purified using the RNA Clean-Up and Concentration Micro Kit. cDNA synthesis was performed using the SuperScript Choice System according to the recommendations of the manufacturer. Using ENZO BioArray HighYield RNA Transcript Labeling Kit, biotin-labeled cRNA was produced. Standard protocol from Affymetrix was used for the in vitro transcription. Quantification of cRNA was performed by spectrophotometric analysis with an A260/A280 ratio of 1.9 to 2.1. Fragmentation of the cRNA was achieved using a defined protocol from Affymetrix. For gene expression profiling, labeled and fragmented cRNA was hybridized to Affymetrix Hugene-2_0-st microarrays with an incubation of 16 h at 45 °C. Affymetrix fluidics station 450 was used to wash the microarrays and scanning was performed with Affymetrix Genechip scanner 3000. A custom CDF version 20 with Entrez-based gene definitions was used to annotate the arrays. Raw fluorescence intensity values were normalized by applying quantile normalization. Differential gene expression analysis was performed with one-way analysis of variance (ANOVA) using the software package JMP10 Genomics version 6 from SAS (SAS Institute). A false positive rate of alpha = 0.05 with FDR correction was taken as the level of significance. The raw and normalized data are deposited in the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/; accession No. GSE121557).
Perfusion of isolated mouse veins
Adult NMRI mice (age: 12-20 weeks, outbred strain) were euthanized and branches of the mesenteric veins were excised, cannulated (with ligated open ends), and inserted into a perfusion chamber (Culture Myograph, DMT, Copenhagen, Denmark), which contained serum-free medium (Panserin 401, PanBiotechTM) supplemented with 50 U/ml penicillin, 50 μg/ml streptomycin, and 0.25 μg/ml Fungizone® antimycotic in the presence of diclofenac (2 µg/ml) or an equivalent volume of a control solvent. The chambers were incubated at 37 °C with 5% CO2, and the veins were cultured for 1 h, (equilibration) followed by exposure to 4 mmHg (control) or 16 mmHg (hypertension) pressure for additional 5 h (occluded at one end). This translates into a fourfold increase in venous wall stress. Vessel segments were processed for whole-mount immunofluorescence analyses. For RNA and protein extraction, several vessel segments of the same experimental group (vessels from one mouse) were pooled.
Quantitative real time RT-PCR (qRT-PCR) analysis
RNA from murine veins was isolated utilizing the RNeasy® Micro Kit 50 (Qiagen, Cat No./ID: 74004), according to the manufacturer’s instructions. Subsequently, cDNA was synthesized using the Sensiscript Reverse Transcription Kit (205213, Qiagen), and quantitative real-time RT-PCR for the target sequences was performed in the Rotor-Gene Q (Qiagen) using the LightCycler® 480 SYBR Green I Master Mix (Roche, Mannheim). Table 1 lists the primer pairs that were utilized for these experiments.
Summary of PCR primer pairs
| Gene name | Sequence |
|---|---|
| S12 | 5’-GAAGCTGCCAAAGCCTTAGA-3’
5’-AACTGCAACCAACCACCTTC-3 |
| Ptgs-1 | 5’-ATGAGTCGAAGGAGTCTCTCG-3’
5’-GCACGGATAGTAACAACAGGGA-3’ |
| Ptgs-2 | 5’-TGAGCAACTATTCCAAACCAGC-3’
5’-GCACGTAGTCTTCGATCACTATC-3’ |
Fluorescence was monitored (excitation at 470 nm and emission at 530 nm) at the end of the annealing phase. Threshold cycle (Ct) was set within the exponential phase of the PCR. Quantification of the PCR product was done by using the ΔΔCt method. Amplification of the ribosomal protein, S12 (S12) cDNA, served as an internal standard.
Capillary electrophoresis
Pressure-exposed veins were lysed in RIPA buffer (pH 7.4) containing 65 mM TRIS, 154 mM NaCl, 10% NP-40, 10% sodium deoxycholate and 1 mM EDTA. Veins were centrifuged at 13,000 × g for 15 min at 4 °C. The supernatant was analyzed by applying capillary electrophoresis due to the low yield of protein. For capillary electrophoresis, the Wes system (ProteinSimple®, San Jose, California, USA) was used. Samples and reagents were prepared according to ProteinSimple® instructions. The primary antibodies for capillary electrophoresis were used using following dilutions: anti-COX-1 (NBP1-85500, Biotechne) 1:20, anti-COX-2 (AF4198-SP, Biotechne) 1:10, anti-ERK1/2 (#4695, Cell Signaling Technology) 1:50, anti-pER1/2 (#4370, Cell Signaling Technology) 1:25, anti-β-actin (ab6276, Abcam) 1:50, anti-MMP2 (NB200-193SS, Biotechne) 1:10. The images were automatically analyzed with Compass for SW software (Version 3.1.7, ProteinSimple®); β-actin and ERK1/2 (for pERK1/2 only) were utilized as references.
Whole-mount immunofluorescence
Vein segments were glued onto a glass slide, fixed with 4% PFA/PBS for 30 min, rinsed in PBST (PBS supplemented with 0.5% Triton X-100) and incubated with the goat anti-CD31 antibody (Biotechne, AF3628; 1:200) and the anti-alpha-SMA-Cy3 (Sigma, C6198; 1:400) for 24 h, rinsed in PBST, incubated with a fluorescein-conjugated secondary antibody (donkey anti-goat, 705-546-147 Dianova; 1:100) for 4 h, rinsed in PBST and counterstained with DAPI for visualization of the nuclei. Vessels were mounted in Mowiol 4-88 mounting media and imaged utilizing a confocal microscope (Olympus IX81, software: xcellence rt 2.0).
Animal models
All animal studies were approved by the Karlsruhe Regional Council and carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No.85-23, revised 1996). Ligation of mouse auricle veins was performed as described previously[17,18]. In brief, male NMRI mice (at least 12 weeks of age) were anesthetized with isoflurane (Baxter, Illinois, USA). Subsequently, one of the three first order veins were ligated using a surgical thread (silk, 7.0, Ethicon, Norderstedt). Remodeling of collateral veins was documented on a daily basis by using a high-resolution digital camera (Digital IXUS 8515, Canon, Tokyo, Japan). High resolution images with a full view of the murine auricle were obtained with a digital camera adjusted to a dissecting microscope (Microscope Wild, Heerbrugg; Leica, Wetzlar, Germany), as described by North and Sanders[17]. Images were then morphometrically analyzed using the software Image J (NIH, Maryland, USA) by measuring across the diameter of the remodeling vessels.
For diclofenac treatment, the auricle was embalmed with 10 µl Voltaren® Spray, which contains 400 µg diclofenac-sodium in liposomes allowing for rapid penetration through the dermis, or ethanol-containing solvent (control), before, one, and three days after ligation of the vein. Four days upon ligation, mice were sacrificed and perfused with Ringer’s solution and a zinc-fixative. Mouse auricles were dissected and processed for paraffin embedding and histological examination.
Morphological analyses of tissue samples
MMP-2, PCNA, COX-2 and CD31 abundance were assessed as a part of the immunofluorescence detection technique performed on 4-5 µm thick paraffin sections of auricle veins by using the corresponding primary antibodies (rabbit anti-MMP2, Biotechne #NB200-193, 1:200; rabbit anti-COX2, CST #4842, 1:200; rabbit anti-PCNA, Abcam #2426, 1:500; goat anti-CD31, Biotechne #AF3628, 1:200) in combination with compatible fluorescence-labeled secondary antibodies (Dianova, 1:100) and employing standard operating procedures. Nuclei were visualized by counterstaining with DAPI.
Statistical analysis
Results are expressed as means ± SD. Differences between two matched experimental groups were analyzed by unpaired Student’s t-test with a probability value of P < 0.05 considered as statistically significant. Differences among 3 or more experimental groups were analyzed by ANOVA, followed by Sidak’s multiple comparisons test.
Results
Biomechanical stress stimulates PTGS2 expression in human venous endothelial cells
Sustained increase in venous intraluminal pressure and consequent increase in circumferential wall tension, may ultimately result in an elevated level of cellular stretch. In order to first assess the various transcriptional targets whose expression may be affected in venous cells under these conditions, HUVECs were exposed to biomechanical stretch for 6 h and processed for RNA extraction. Subsequent gene expression profiling [Figure 1, Supplement 1] revealed significant up-regulation of transcripts that control inflammation (e.g., CXCL8, ESM-1, VCAM-1), growth/differentiation (KITLG/KIT, TGFB2, PDGFC, HBEGF, GDF6) and extracellular matrix remodeling (HAS2, ADAMTS2). Down-regulated transcripts included those associated with nitric-oxide signaling (NOS3, guanylate cyclases), redox-signaling (HMOX1, TXNIP) and transcriptional modulation of cellular differentiation (SMAD6, KLF2, SNAI2).
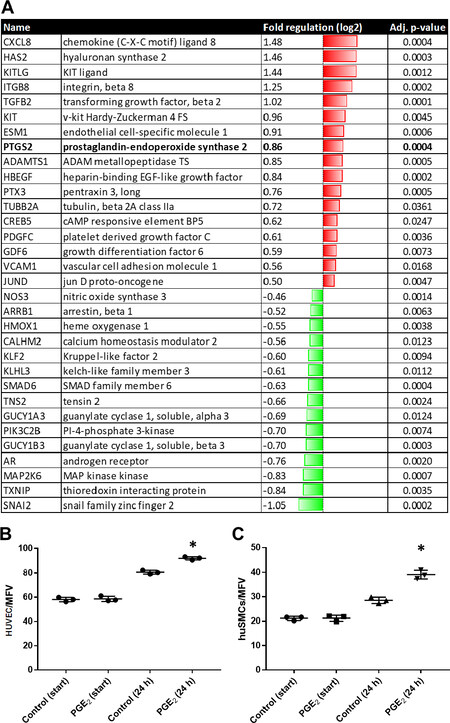
Figure 1. Transcriptional profile of stretch-stimulated HUVECs (A); responses of HUVECs (B); and huSMCs (C) to prostaglandin E2. HUVECs from three different donors were subjected to biomechanical stretch (6 h). mRNA was isolated and processed for microarray-based analysis. Table A shows selected genes which were affected by biomechanical stretch (vs. static control) based on the following criteria: fold change (log2) > 0.5 (rounded up), P < 0.05 for adjusted P-value (ANOVA), n = 3; green: down-regulated, red: up-regulated. Only significantly regulated transcripts were listed; red: elevated transcript level; green: decreased transcript level. For detailed expression data, see Gene Expression Omnibus (GEO): GSE121557. HUVECs and huSMCs were stimulated with prostaglandin E2 (10 µM) for 24 h. The number of cells in five randomly selected microscopic fields of view (MFV) was determined every day (B and C, *P < 0.05 vs. control, n = 3).
PTGS2 encoding COX-2 was identified as another significantly up-regulated transcript in stretch-stimulated HUVEC [Figure 1A], confirming results from earlier publications[15]. COX-2 attracted our attention as it encodes a stress-inducible enzyme that synthesizes prostaglandin H2 (PGH2), thus acting as a precursor molecule for multiple prostanoid effectors capable of activating vascular cells. For instance, stimulation with PGE2 spurred the proliferation of both endothelial as well as vascular smooth muscle cells [Figure 1B and C, Supplement 2]. Moreover, COX-2 has been associated with the development of varicose veins and is an important drug target for the non-steroidal anti-inflammatory drug class (NSAIDs). Currently, it is still unknown whether COX activity may causally contribute to the remodeling of the venous wall or represents a mere consequence of the cellular activation under these conditions.
Diclofenac decreases pressure-induced ERK1/2 and MMP-2 activation in mouse veins
In order to better elucidate the role of COX in varicose development, we aimed to investigate its functional relevance in the activation of biomechanically stressed venous cells. To this end, we developed an experimental model that allows exposing mouse veins to elevated pressure levels while being treated with diclofenac-an inhibitor of COX-1 and COX-2 activity. Venous wall stress was elevated by increasing the pressure from 4 (physiological pressure level) to 16 mmHg (mimics venous hypertension), which by itself, had no major impact on the architecture and integrity of the venous wall as evidenced by whole-mount immunofluorescence analyses [Figure 2A]. However, nanoscale protein analyses performed by applying capillary electrophoresis, indicated a pressure-induced increase in COX-2, but not COX-1 protein abundance in venous cells [Figure 2B-G]. In contrast, the expression of the corresponding genes Ptgs1/2 was not significantly altered under these conditions [Supplement 3]. Corresponding experiments with diclofenac-treated veins showed a decline in the protein level of COX-2, but not of COX-1 [Supplement 4].
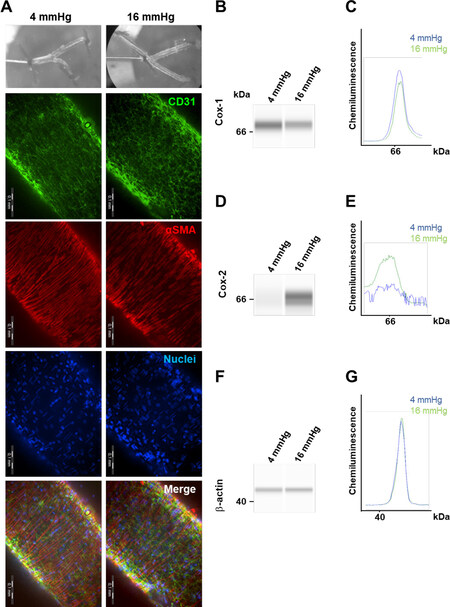
Figure 2. Pressure stimulation of isolated mouse veins. Isolated branches of the mesenteric vein of male NMRI mice were exposed to pressure levels of 4 or 16 mmHg, respectively. Afterwards, vessel segments were processed for whole mount immunofluorescence staining, scale bars: 0.1 mm (A) to detect CD31 (endothelial cell marker), αSMA (smooth muscle cell marker), and DAPI (visualization of the nuclei). Protein samples generated from these blood vessels were pooled and analyzed by automated capillary electrophoresis/immunodetection [required due to the low yield of protein; specified antigens were detected by antibodies and corresponding signals (B, D, F) and were automatically evaluated by their size (kDa) and intensity (area under the curve, C, E, G)]. Signals specific for COX-1 (B and C) and COX-2 (D and E) were detected. b-actin served as a loading reference (F and G).
To evaluate the cellular activity during this early phase of biomechanical stress response, we determined the levels of phosphorylation of the MAP-kinases ERK1/2 in the venous wall, a known and important indicator of growth regulating signaling cascades. Additionally, we assessed the abundance of the pro-form (~ 72 kDa) and active form (~ 62 kDa) of the matrix-metalloproteinase, MMP-2 [Supplement 5], a marker of proteolytic capacity of venous cells. Exposure to 16 mmHg intraluminal pressure led to an increased level of ERK1/2 phosphorylation [Figure 3A] and an abundance of active MMP-2 in the venous wall [Figure 3B]. Diclofenac treatment diminished these effects [Figure 3A and B].
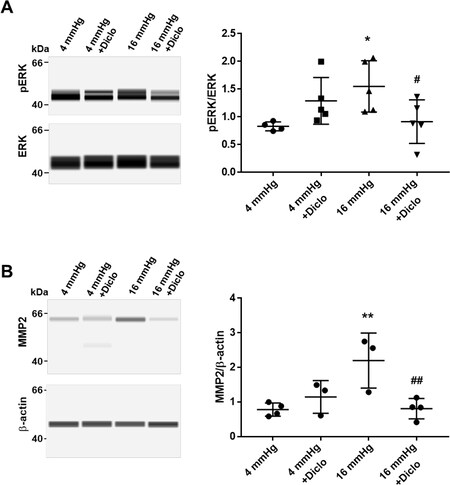
Figure 3. Impact of elevated pressure levels on markers of cell activity. Isolated branches of the mesenteric vein of male NMRI mice were exposed to 4- or 16-mmHg, respectively in the presence or absence of diclofenac. Protein extracts generated from these blood vessels were analyzed by capillary electrophoresis as described above. The level of phosphorylated ERK1/2 and the abundance of MMP-2 (pro-enzyme: 72 kDa was below detection limit, active form: ~ 62 kDa) were determined; A: *P < 0.05 vs. 4 mmHg, #P < 0.05 vs. 16 mmHg, n = 4-5 as indicated; B: **P < 0.01 vs. 4 mmHg, ##P < 0.01 vs. 16 mmHg, n = 3-4 as indicated.
Diclofenac inhibits varicose-like venous remodeling in mice
The results obtained thus far suggested that diclofenac may inhibit, or at least delay, pressure-induced or biomechanically evoked stress responses of venous cells. Consequently, we hypothesized that treatment with diclofenac will interfere with the onset of varicose-like remodeling in mammalian veins, in vivo. To test this, we employed a mouse model, in which varicose-like venous remodeling can be mimicked by occlusion of a single vein in the mouse auricle to increase the filling pressure in the connected local venous network[5,17-20]. Diclofenac was applied transdermally by utilizing liposome-based diclofenac formulation (Voltaren® spray), and venous remodeling was determined by comparing the diameter of veins immediately and four days after the venous occlusion.
While a corkscrew-like morphology and a significant increase in the diameter of remodeling veins was observed under control conditions, this effect was significantly attenuated upon treatment with diclofenac [Figure 4A]. Immunofluorescence-based techniques indicated a generally low COX-2 protein abundance in the unaffected auricle veins [Figure 4B and C], but it was predominantly localized in endothelial cells of remodeling vessels [arrows, Figure 4D and E]. Further analyses detected the proliferation marker, PCNA, the protease MMP-2, and COX-2 in cells of enlarged (remodeled) veins [Figures 5A-C], thus confirming earlier observations[5,17-20]. These markers were barely detectable in vessels from auricles treated with diclofenac [Figures 5D-F].
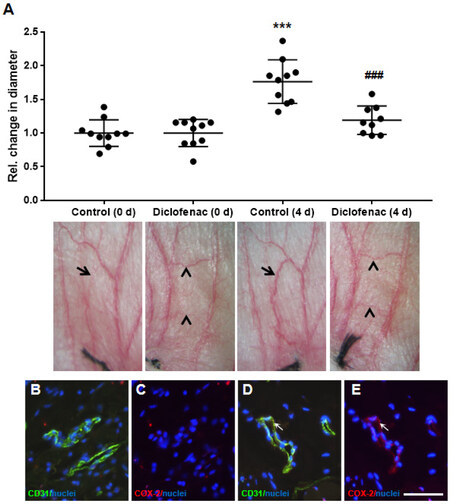
Figure 4. Effect of diclofenac on venous remodeling, in vivo. Imaging of the mice auricular vein vasculature immediately after and 4 days after ligation of the auricle vein revealed an increase in tortuosity and diameter of veins connected to the occluded venous network (arrow in A, ***P < 0.001 vs. control 0 d, n = 10). This was significantly inhibited (arrowheads in A, ##P < 0.01 vs. control 4 d, n = 10) by local administration of diclofenac (Voltaren Spray®). Control auricles were treated with an inert solvent. Original diameter of the remodeling veins was set to 1. Mouse auricle tissue (control, 4 d) was fixed, paraffin-embedded and cross-sections of veins were analyzed by immunofluorescence staining to detect CD31 (endothelial cell marker) and COX-2 in combination with DAPI. COX-2 was barely detected in veins undergoing no remodeling process (B and C), but was detectable in endothelial cells of remodeling veins (arrows in D and E, scale bar: 50 µm).
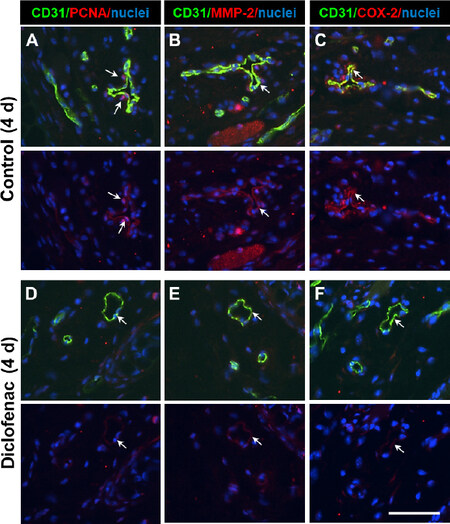
Figure 5. Influence of diclofenac-treatment on SMC activation in mouse auricle veins. Selected auricle tissue segments (4 d after vein ligation) containing remodeling veins were fixed, paraffin-embedded, cross-sectioned, and processed for immunostaining (nuclei were visualized by DAPI staining). While PCNA (proliferation marker, A, arrows), MMP-2 (B, arrows), and COX-2 (C, arrows) were detectable in cells of the remodeling veins, corresponding proteins were barely detected in diclofenac-treated auricles (arrows in D-F, scale bar: 50 µm).
Discussion
Diclofenac belongs to the class of non-steroidal anti-inflammatory drugs (NSAIDs), which are broadly applied to fight fever, attenuate pain, prevent thrombosis, or limit inflammatory responses. NSAIDs specifically inhibit the activity of COX-1 and COX-2 that act as rate-limiting factors in prostanoid synthesis by generating prostaglandin H2 (PGH2) from arachidonic acid. Depending on the cell-specific availability of defined prostaglandin synthases (e.g., prostaglandin E2 synthase or prostaglandin I2 synthase), several bioactive products such as PGE2 and prostacyclin (PGI2) may be generated from PGH2. In the vascular system, recent findings suggest that COX-1 and PGI2-synthase are constitutively active in endothelial, but not vascular smooth muscle cells[21,22], although COX-1 is also detected in murine vascular smooth muscle cells in vivo[23]. In contrast, induction of vascular COX-2/PTGS-2 expression is usually associated with inflammatory responses[24], atherosclerosis[25], or responses to injury[23]; but it may additionally be constitutively expressed in distinct organs such as the kidney[26].
In this study, we have verified both COX-1 and COX-2 protein expression in isolated and cannulated branches of mesenteric mouse veins whose RNA expression was slightly, but not significantly increased after elevating the intraluminal pressure for several hours. In vivo, COX-2 was barely detectable in quiescent auricle veins but was abundant in endothelial cells of remodeling veins. Although the regulatory determinants remain obscure, upregulation of COX-2 under these conditions correlates with the induction of biomechanical stress, which is shown to drive COX-2 expression in human endothelial cells in this and prior studies[15]. However, while the presence of COX may further result in the production of PGH2, the release of defined prostanoids and the outcome of the corresponding signaling events are controlled by cell-specific distribution of terminal prostaglandin-synthases and prostaglandin-receptors.
In the vascular system, PGI2 as well as PGE2 are crucial for regulating vascular tone and inflammatory responses via specific G-protein coupled receptors located on the surface of vascular endothelial and smooth muscle cells. Indeed, PGI2 acts as the most relevant cardioprotective prostanoid[21]. In line with this, mutations/polymorphisms in the human prostacyclin synthase gene have been associated with stroke, myocardial infarction, and essential hypertension[27]. In vascular smooth muscle cells, the prostacyclin (IP) receptor is usually coupled to the G-protein subunit Gαs that is connected to a signaling cascade, which promotes relaxation and a decrease in vascular tone[28]. Consequently, prostacyclin synthase knockout mice develop hypertension[29]. For PGE2, the signaling pathways become even more complex, since PGE2 may activate at least four different receptors (EP1-EP4) coupled to several subtypes of G-proteins, which drive multiple stimulatory as well as inhibitory effects in vascular cells[30]. For instance, activation of EP receptors in endothelial cells has been shown to trigger pro-inflammatory responses, MMP-9 expression, and angiogenesis[30]. In line with these observations, injury-induced arterial hyperplasia appears to be controlled through the COX-2-PGE2-EP3 signaling axis[23], while knockout of microsomal prostaglandin E2 synthase-1 inhibits this process[31].
Although supplementary corresponding analyses are lacking for venous diseases, the association of varicose vein development with accentuated COX expression[14], increased PGE2 levels and decreased PGI2 levels[13] have been observed previously. Under these conditions, diclofenac may interfere with the activity of COX, with a preference for COX-2 due to its pharmacological mechanism of action[32]. As a consequence, lower PGI2 and PGE2 levels would result in dampened EP-receptor-mediated signaling that is closely connected with MAP kinases. This relationship may explain the reduced ERK1/2 phosphorylation in pressure-exposed diclofenac-treated veins indicating a diminished cellular activation that is also reflected by the lower abundance of active MMP-2. Consequently, in vivo, diclofenac may hamper the stress-induced proliferation and proteolytic capacity of venous cells, attenuating or delaying varicose-like venous remodeling, as evidenced in this study.
One of the limiting factors for the translational impact of our study is the utility of a mouse model, which only mimics certain aspects of venous remodeling processes in humans. As such, although occlusion- or reflux-mediated venous wall stresses induce comparable cellular responses in mice and humans[5,18], and venous harvesting and conduits in mice bear resemblance with venous diseases in men; their size, structure, and overall rate of remodeling nevertheless differ significantly. Moreover, small animals are not generally exposed to drastic changes in hydrostatic pressure levels even if they shortly stay in an erect position. For instance, normal supine venous pressures of 5-10 mmHg in humans are comparable to the values measured in mice (~ 6 mmHg); and under experimental conditions, the pressure in mouse veins may increase three- to four-fold. In contrast, in the human venous vasculature, pressure levels of up to 24 mmHg in the supine position and up to 120 mmHg in the erect posture, have been measured in association with venous diseases[33].
On the molecular level however, many cellular responses in the remodeling veins are comparable between the two species, considering their proteolytic, transcriptional, or proliferative activity[5,6,17,18]. In mice, these parameters are influenced directly by the increase in the wall stress (thus indicating its relevance as a trigger for venous remodeling); in humans, analysis is usually focused on veins that are in later stages of the venous diseases progression, where the aforementioned parameters may also be affected by additional determinants such as inflammatory processes. As our mouse model does not mimic aspects associated with late-stage venous diseases progression, the impact of diclofenac on venous inflammation and edema formation, specifically, would be better addressed in another context. However, considering its broad spectrum of activity, inhibition of COX by diclofenac may indeed provide an additional benefit in attenuating inflammatory responses as well as the associated sensations of pain, which is characteristic of venous diseases as has been shown for acute varicophlebitis or acute superficial thrombophlebitis of the lower limbs[34]. Although the influence of diclofenac on early stages of venous remodeling or diseases has not been analyzed in detail, it may diminish not only inflammation and thrombosis, but may also dampen the prostanoid-mediated proteolytic activity and proliferation in the vessel wall, which slowly and steadily has the potential to spur further venous disease development.
Declarations
AcknowledgmentsThe authors would like to acknowledge Ralph Mayer and Dr. Caroline Arnold for their technical assistance in this study.
Authors’ contributionsPerformed experiments, analyzed the data, and revised the manuscript: Lust L, Kuk H, Kohlhaas J
Performed the microarray analysis and the statistical evaluation: Sticht C
Designed the study, analyzed the data and wrote the manuscript: Korff T
Availability of data and materialsThe raw and normalized data are deposited in the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/; accession No. GSE121557).
Financial support and sponsorshipThis work was supported by the Swiss Society for Phlebology.
Conflicts of interestAll authors declare that there are no conflicts of interest.
Ethical approval and consent to participateHuman umbilical vein endothelial cells (HUVECs) were isolated as per approval from the Local Ethical Committee (document number 336/2005, Heidelberg Germany) and conformed to the principles outlined in the Declaration of Helsinki (1997). Parental consent was obtained for isolation of HUVECs from the umbilical cords of newborns.
Consent for publicationNot applicable
Copyright© The Author(s) 2021.
REFERENCES
1. Beebe-Dimmer JL, Pfeifer JR, Engle JS, Schottenfeld D. The epidemiology of chronic venous insufficiency and varicose veins. Ann Epidemiol 2005;15:175-84.
2. Burkitt DP, Townsend AJ, Patel K, Skaug K. Varicose veins in developing countries. Lancet 1976;2:202-3.
3. Lee AJ, Evans CJ, Allan PL, Ruckley CV, Fowkes FG. Lifestyle factors and the risk of varicose veins: Edinburgh Vein Study. J Clin Epidemiol 2003;56:171-9.
5. Feldner A, Otto H, Rewerk S, Hecker M, Korff T. Experimental hypertension triggers varicosis-like maladaptive venous remodeling through activator protein-1. FASEB J 2011;25:3613-21.
6. Pfisterer L, König G, Hecker M, Korff T. Pathogenesis of varicose veins-lessons from biomechanics. Vasa 2014;43:88-99.
7. Ligi D, Croce L, Mannello F. Chronic venous disorders: the dangerous, the good, and the diverse. Int J Mol Sci 2018;19:2544.
8. Wali MA, Eid RA. Changes of elastic and collagen fibers in varicose veins. Int Angiol 2002;21:337-43.
10. Sansilvestri-Morel P, Rupin A, Jaisson S, Fabiani JN, Verbeuren TJ, Vanhoutte PM. Synthesis of collagen is dysregulated in cultured fibroblasts derived from skin of subjects with varicose veins as it is in venous smooth muscle cells. Circulation 2002;106:479-83.
12. Gomez I, Benyahia C, Louedec L, et al. Decreased PGE2 content reduces MMP-1 activity and consequently increases collagen density in human varicose vein. PLoS One 2014;9:e88021.
13. Biagi G, Lapilli A, Zendron R, Piccinni L, Coccheri S. Prostanoid production in varicose veins: evidence for decreased prostacyclin with increased thromboxane A2 and prostaglandin E2 formation. Angiology 1988;39:1036-42.
14. Bertrand-Thiebault C, Ferrari L, Boutherin-Falson O, et al. Cytochromes P450 are differently expressed in normal and varicose human saphenous veins: linkage with varicosis. Clin Exp Pharmacol Physiol 2004;31:295-301.
15. Zhao H, Hiroi T, Hansen BS, Rade JJ. Cyclic stretch induces cyclooxygenase-2 gene expression in vascular endothelial cells via activation of nuclear factor kappa-beta. Biochem Biophys Res Commun 2009;389:599-601.
16. Avendaño MS, Martínez-Revelles S, Aguado A, et al. Role of COX-2-derived PGE2 on vascular stiffness and function in hypertension. Br J Pharmacol 2016;173:1541-55.
17. Pfisterer L, Meyer R, Feldner A, Drews O, Hecker M, Korff T. Bortezomib protects from varicose-like venous remodeling. FASEB J 2014;28:3518-27.
18. Eschrich J, Meyer R, Kuk H, Wagner AH, Noppeney T, Debus S, et al. Varicose remodeling of veins is suppressed by 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. J Am Heart Assoc 2016;5:e002405.
19. Kuk H, Arnold C, Meyer R, Hecker M, Korff T. Magnolol inhibits venous remodeling in mice. Sci Rep 2017;7:17820.
20. Kuk H, Arnold C, Wagner AH, Hecker M, Sticht C, Korff T. Glycyrrhetinic acid antagonizes pressure-induced venous remodeling in mice. Front Physiol 2018;9:320.
21. Mitchell JA, Kirkby NS. Eicosanoids, prostacyclin and cyclooxygenase in the cardiovascular system. Br J Pharmacol 2019;176:1038-50.
22. Kirkby NS, Lundberg MH, Harrington LS, et al. Cyclooxygenase-1, not cyclooxygenase-2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc Natl Acad Sci U S A 2012;109:17597-602.
23. Zhang J, Zou F, Tang J, et al. Cyclooxygenase-2-derived prostaglandin E2 promotes injury-induced vascular neointimal hyperplasia through the E-prostanoid 3 receptor. Circ Res 2013;113:104-14.
24. Jones DA, Carlton DP, McIntyre TM, Zimmerman GA, Prescott SM. Molecular cloning of human prostaglandin endoperoxide synthase type II and demonstration of expression in response to cytokines. J Biol Chem 1993;268:9049-54.
25. Baker CS, Hall RJ, Evans TJ, et al. Cyclooxygenase-2 is widely expressed in atherosclerotic lesions affecting native and transplanted human coronary arteries and colocalizes with inducible nitric oxide synthase and nitrotyrosine particularly in macrophages. Arterioscler Thromb Vasc Biol 1999;19:646-55.
26. Kirkby NS, Chan MV, Zaiss AK, et al. Systematic study of constitutive cyclooxygenase-2 expression: Role of NF-kappaB and NFAT transcriptional pathways. Proc Natl Acad Sci U S A 2016;113:434-9.
27. Nakayama T. Genetic polymorphisms of prostacyclin synthase gene and cardiovascular disease. Int Angiol 2010;29(2 Suppl):33-42.
28. Midgett C, Stitham J, Martin K, Hwa J. Prostacyclin receptor regulation--from transcription to trafficking. Curr Mol Med 2011;11:517-28.
29. Yokoyama C, Yabuki T, Shimonishi M, et al. Prostacyclin-deficient mice develop ischemic renal disorders, including nephrosclerosis and renal infarction. Circulation 2002;106:2397-403.
30. Foudi N, Gomez I, Benyahia C, Longrois D, Norel X. Prostaglandin E2 receptor subtypes in human blood and vascular cells. Eur J Pharmacol 2012;695:1-6.
31. Wang M, Ihida-Stansbury K, Kothapalli D, et al. Microsomal prostaglandin e2 synthase-1 modulates the response to vascular injury. Circulation 2011;123:631-9.
32. Patrono C, Patrignani P, García Rodríguez LA. Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. J Clin Invest 2001;108:7-13.
33. Raju S, Knight A, Lamanilao L, Pace N, Jones T. Peripheral venous hypertension in chronic venous disease. J Vasc Surg Venous Lymphat Disord 2019;7:706-14.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lust L, Kuk H, Kohlhaas J, Sticht C, Korff T. Inhibition of cyclooxygenase activity by diclofenac attenuates varicose remodeling of mouse veins. Vessel Plus 2021;5:7. http://dx.doi.org/10.20517/2574-1209.2020.52
AMA Style
Lust L, Kuk H, Kohlhaas J, Sticht C, Korff T. Inhibition of cyclooxygenase activity by diclofenac attenuates varicose remodeling of mouse veins. Vessel Plus. 2021; 5: 7. http://dx.doi.org/10.20517/2574-1209.2020.52
Chicago/Turabian Style
Lust, Leandra, Hanna Kuk, Johanna Kohlhaas, Carsten Sticht, Thomas Korff. 2021. "Inhibition of cyclooxygenase activity by diclofenac attenuates varicose remodeling of mouse veins" Vessel Plus. 5: 7. http://dx.doi.org/10.20517/2574-1209.2020.52
ACS Style
Lust, L.; Kuk H.; Kohlhaas J.; Sticht C.; Korff T. Inhibition of cyclooxygenase activity by diclofenac attenuates varicose remodeling of mouse veins. Vessel Plus. 2021, 5, 7. http://dx.doi.org/10.20517/2574-1209.2020.52
About This Article
Copyright

Data & Comments
Data
0


 Cite This Article 6 clicks
Cite This Article 6 clicks

 Like This Article 6
likes
Like This Article 6
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.