
Oxidative stress and heart disease: the thyroid hormone mediation

Abstract
The heart is one of the principal targets of thyroid hormones (TH) action, affecting cardiac contractility, heart rate, and diastolic function. Oxidative damage is pivotal for onset and development of cardiovascular disease (CVD) and heart failure. Specifically, free radical generation is associated to hyper-metabolic state in hyperthyroidism, whereas hypo-metabolic state induced by hypothyroidism leads to a decrease of oxidative stress. In the present review, the role of oxidative damage in CVD and failing heart-TH interplay will be considered. The main oxidative events leading to cardiac dysfunction and TH cardiac regulation at genomic and non-genomic levels will be discussed, as well as role of TH in cardioprotection and reversion of cardiac remodeling.
Keywords
Introduction
The heart is one of the principal targets of thyroid hormones (TH) action, in fact, any change in TH directly or indirectly affects the cardiac metabolism, influencing contractility, heart rate, and diastolic function. Oxidative damage is one of the most relevant mechanisms responsible for the progression of cardiovascular disease (CVD) and heart failure[1,2]. Furthermore, free radical production is associated to hyper-metabolic state in hyperthyroidism, whereas the hypo-metabolic state induced by hypothyroidism leads to a decrease in free radical production[3-5]. In the present review, the role of oxidative damage in CVD and failing heart-TH interplay will be considered. The main oxidative events leading to cardiac disease and compromise will be described and TH cardiac regulation at genomic and non-genomic levels will be evaluated. Finally, TH importance in cardioprotection and reversion of cardiac remodeling will be discussed.
Oxidative stress and heart: an overview
A correct functioning of the cardiac muscle requires a constant supply of oxygen, in order to ensure an efficient cellular metabolism. In the last decades, many clinical and experimental studies have been focused on the critical role of oxidative stress (OxS) in several cardiac derangements[6]. OxS is defined as the imbalance between reactive oxygen species (ROS) with respect to the antioxidant defense system[7]. ROS are physiologically involved in many biological processes since they act as second messengers in different cellular pathways. However, in pathological conditions, ROS overproduction can become extremely dangerous to proteins, membranes, and nucleic acids, thus compromising crucial enzymes and causing mutagenesis at DNA level[7-9]. ROS induction in the heart is promoted by a number of mechanism involving nicotinamide adenine dinucleotide phosphate (NADPH), xanthine oxidases (XO), cytochrome P450 (CYP450), mitochondrial production, uncoupling of endothelial nitric oxide synthetase (eNOs), and auto-oxidation of catecholamines[6]. OxS is often triggered by the complex interactions among the different pro-oxidant systems. Moreover, the concomitant inflammatory circuit progression may further sharpen the risk of cardiovascular disease[10]. In the vessel wall, NADPH oxidase is the principal source of ROS and, in mammals, there are seven different types of NADPH oxidases, all containing one catalytic subunit among Nox1-Nox5, DUOX1, DUOX2, and up to five regulatory subunits. Different stimuli may up-regulate Nox isoforms with the consequent increase of ROS. The interplay between NADPH oxidase and mitochondria leads to the reciprocal stimulation of ROS production[11]. Generated ROS, in turn, promote and maintain XO activity and superoxide anion O2-· synthesis, at the expenses of NO, further triggering new ROS synthesis[12]. At the cardiac level, superoxide anion radical O2-·, hydroxyl radical ·OH, and hydrogen peroxide H2O2 are considered the most important ROS and are involved in the hypertrophy, myocardial growth, fibroblast proliferation, and cardiac remodeling, and reduction of cardiac function. The action of most ROS is restricted to the intracellular environment; however, H2O2 species have higher diffusion capacity through the cell membrane, reaching more distant targets[13]. On the antioxidant counterpart, protective mechanisms balancing the dangerous effects of ROS include the scavenging activity of superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GSHPx)[2]. In particular, the 3 SOD isoforms have crucial antioxidant activities, catalyzing the dismutation reaction from O2- to O2 and H2O2. The three isoforms are located at different cellular sites, SOD1 is in the cytoplasm and in the intermembrane space of mitochondrion, SOD2 in the mitochondrial matrix, whereas SOD3 is called “extracellular SOD” because of its location in the extracellular matrix[14]. SOD enzymes have an important anti-atherosclerotic function whose efficacy strongly depends on downstream CAT and GSHPx-mediated H2O2 detoxification. The most relevant consequence of OxS is the damage of nucleic acids, cell membrane lipids, proteins, and enzymes, provoking severe compromise of cellular response and generation of secondary reactive species. Finally, when ROS generation overwhelms the antioxidant effectiveness of the cell, inevitably leads to cell death by necrosis or apoptosis. A schematic representation of main cellular sources of reactive oxygen species (ROS) activated in heart disease and the antioxidant counterpart is represented in Figure 1.
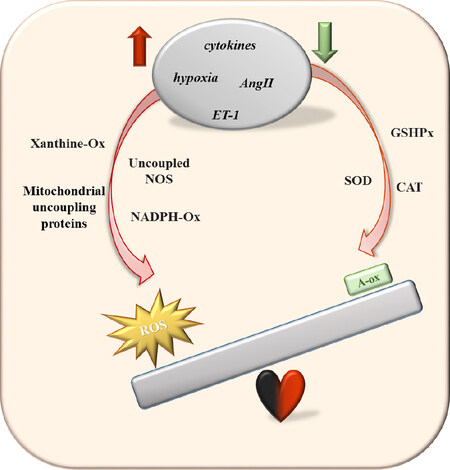
Figure 1. Schematic representation of main cellular sources of ROS activated in heart disease and the antioxidant counterpart. ROS: reactive oxygen species; AngII: angiotensin II; ET-1: endothelin-1; Ox: Oxidase; NOS: nitric oxide synthase; NADPH: nicotinamide adenine dinucleotide phosphate; SOD: superoxide dismutase; GSHPx: glutathione peroxidase; CAT: catalase
Thyroid hormones and heart: general aspects
Triiodothyronine (T3) and thyroxine (T4) produced by the thyroid have multiple effects on the heart. Circulating T4 derives from the thyroid, whereas T3 is produced in the peripheral tissues by T4 5’-monodeiodination. TH affect both diastolic and systolic performances and have important effects on cardiac growth and differentiation, morphology, cell protection, and metabolism[15,16]. TH genomic action on the heart can be mediated by specific nuclear TH receptors and regulate specific cardiac gene expression. There are many cardiac genes considered as targets of transcriptional stimulation by TH. The principal factor regulating cardiac muscle contraction/relaxation activity is intracellular free Ca2+ concentration, which is regulated by the synergistic interaction between Ca2+ release from the sarcoplasmic reticulum through the ryanodine receptors and Ca2+ re-uptake through the sarcoplasmic reticulum calcium-activated ATPase (SERCA2)[17]. SERCA2 activity, in turn, is regulated by phospholamban, an integral sarcolemma protein modulated by protein kinases via phosphorylation which has maximal inhibitory activity when dephosphorylated[18]. T3 positive activation of SERCA2 and negative regulation of phospholamban, determine an accelerated sarcoplasmic reticulum Ca2+ re-uptake and subsequent cardiomyocyte relaxation of diastole, whereas the opposite occurs during the systolic phase[19]. The hypothyroid condition is characterized by an impairment of Ca2+ intracellular loading and improvement of systolic function. Furthermore, TH have opposite effects on gene expression of myosin heavy chains MHC-α and MHC-β in the heart since they activate MHC-α and inhibit MHC-β. The most efficient cardiac myosin isoform (V1) is composed by two MHC-α subunits and has a more rapid shortening velocity than the other less efficient isoenzymes (V2 and V3), in which one or both MHC subunits are of β type. T3 induces a rapid switch from MHC-β toward MHC-α, leading to an increased shortening velocity in ventricle myocardium[20]. Moreover, TH can act also via non-genomic activity, throughout the interaction with cytoplasmic and membrane-associated receptors, in order to maintain a cardiovascular homeostasis by reaching a high level of integration with inflammatory, neuroendocrine systems and the oxidative stress machinery[15]. The non-genomic effects of TH are independent from nuclear uptake of TH and seem to be mediated by receptors structurally analogous to TH receptors, in particular by the truncated isoforms of TRα, called holoreceptor, and located at plasma membrane, cytoplasm, and mitochondria[21]. Furthermore, structurally unrelated receptors have been considered involved in the non-genomic mediation of TH effects in the target cells, such as the plasma membrane integrin αvβ3[22]. Cardiac non-genomic effects of TH are mainly referred to the TH modulation of ion transporters and channels such as Na+/K+ activated ATPases, Na+/Ca2+ exchanger, and voltage-gated K+ channel isoforms (Kv4.2 and Kv4.3), mediating the Ca2+-independent transient outward current, whose role is to determine the electrochemical behavior of the myocardium[23,24].
Oxidative stress, coronary artery disease and myocardial infarction
Enhanced generation of ROS and reactive nitrogen species in the heart induce a higher consumption of oxidants and a strong reduction of antioxidant enzymes, driving to an OxS condition. The heart is a constantly active organ and needs an adequate supply of oxygen and nutrients from the blood stream. Therefore, structural/functional derangements, acting at different levels in the cardiovascular system (e.g., coronaries, myocardium, heart valves, blood pressure, etc.), may increase the risk of heart failure. Alteration of oxidative equilibrium may be due either to the overproduction of ROS or to the inhibition of scavenging mechanisms. Inflammatory response associated to atherosclerotic events (i.e., endothelial dysfunction and hyperlipidemia) and myocardial compromise (i.e., heart failure and myocardial infarction) are considered major inducers of OxS.
Coronary artery disease is the number one cause of mortality worldwide and occurs when changes in the artery wall lead to the accumulation of atherosclerotic plaques[25]. During coronary inflammatory outbreak, OxS plays a crucial role in the pathological LDL oxidation, NO reduction, monocyte and macrophage overproduction, and accumulation of metalloproteinases in vascular endothelium. OxS-induced growth and proliferation of vascular muscle cells also cause an excess of growth factors (e.g., fibroblast growth factor and its receptor, insulin growth factor and its receptor, etc.) and adhesion molecules [e.g., intercellular adhesion molecule (ICAM-1), platelet adhesion molecule (PECAM-1), vascular cell adhesion molecule (VCAM-1)][26-28]. Atherosclerosis plaque narrows the vessel lumen and restricts blood flow progressively leading to ischemic events and oxygen shortage. Complete blockage of blood flow, frequently via a thrombotic event, induces a myocardial infarction[29]. Inflammatory response at the infarct region and associated neuro-hormonal activation determine a serious modification in myocardial metabolism[30]. Mitochondrial unbalancing in ischemia reperfusion is further worsen by intracellular Ca2+ overload caused by the opening of voltage-sensitive Ca2+ channels and membrane ions exchangers with a Ca2+ and Na2+ accumulation inside the cells[31]. OxS exacerbation is associated to mitochondrial impairment of electron chain transport activity, inflammatory cytokines accumulation, and extracellular matrix production, in the pathological attempt to repair damaged cardiac tissue[32,33]. After the infarct of myocardium, a relevant loss of functional tissue occurs and the following modification in structure, geometry and metabolism of the heart is called cardiac remodeling[34]. Main events associated to cardiac remodeling are represented in Figure 2. Cardiac remodeling is a multifactorial process and regards many aspects strictly inter-related such as oxidative stress promotion, inflammatory cytokines, and metalloproteinases activation, mechanical stress, hypertrophy, apoptosis, and necrosis. The final common result is the loss of contractile capacity of the still vital myocardium in a compensatory effort, aiming to maintain a sufficient perfusion pressure in the heart. Even though in the initial phase, all the occurring changes seem to be compensatory, successively they turn out to be maladaptive and unbeneficial. At the cellular level, a characteristic marker of cardiac remodeling consists in the so-called fetal pattern reactivation, a low-energy state that tries to adapt and protect the damaged myocardium upon stress[35]. The features of fetal heart metabolism re-emerged, glucose metabolism for energy providing is favored with respect to fatty acids substrates and the a-MHC to b-MHC switch takes place, typical for the progression of the pathological cardiac hypertrophy observed in heart failure.
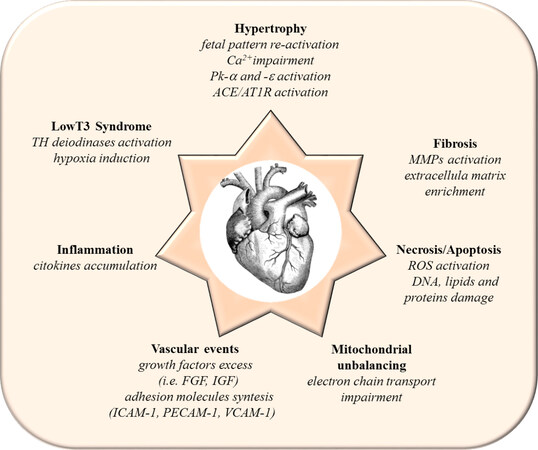
Figure 2. Main events associated to cardiac remodeling. Pk: protein kinase; ACE: angiotensin converting enzyme; AT1R: angiotensin type I receptor; TH: thyroid hormones; MMPs: metallo proteinases; ROS: reactive oxygen species; FGF: fibroblast growth factor; IGF: insulin growth factor; ICAM-1: intercellular adhesion molecule-1; PECAM-1: platelet adhesion molecule-1; VCAM-1 vascular cell adhesion molecule-1
Oxidative stress, thyroid hormones and cardioprotection
Altered cardiac metabolism in the failing heart is related to unbalanced myocardial contractile function, compromised cell signaling, mitochondrial impairment, and exacerbation of inflammatory machinery[1]. In this pathological picture, all the mechanisms contributing to reduce or prevent myocardial injury are considered part of the cardioprotection process. Therefore, any possible protective factor might be potentially investigated with the final aim to develop novel therapeutical strategies to contrast post-ischemic heart failure evolution. Among new treatments to combat reperfusion injury and cardiac remodeling, TH showed a good efficacy in contrasting OxS detrimental products and carry out a reparative action on the diseased myocardium[36,37]. It has been long recognized that normal thyroid homeostasis can vary in particularly stressful conditions such as myocardial infarction and several organ compromise, and that in this condition the fall in circulating T3 and a rise in reverse (rT3) are evident, whereas T4 and thyroid-stimulating hormone (TSH) usually remain normal, even though T4 levels can drop as well in the severely ill patients. This state is called low T3 syndrome (LT3S)[38,39]. The most reliable pathophysiological mechanism to explain LT3S are OxS and hypoxia induction, reduction of 5’-monodeiodinase (DIO1 and DIO2) enzyme activity, and activation of 5-monodeiodination (DIO3) enzyme[40,41]. The decrease of TH activating enzymes DIO1 and DIO2, and the rise of TH inactivating enzyme DIO3 are observed in infarcted myocardium. The deiodinases arrangement induce a local hypothyroid state in the remodeling ventricle, with the final decrease of active T3 and T4 and the increase of inactive rT3. Furthermore, accumulating evidence shows that the LT3S is a strong prognostic, independent predictor of death in patients affected by both acute and chronic heart disease. In experimental models of cardiac hypertrophy or myocardial infarction, alterations in the TH signaling, concerning cardiac mitochondrion, interstitium, and vasculature, have been suggested to be strictly related to heart dysfunction[15]. In fact, TH regulate many genes known to have a cardioprotective and antioxidant action with antiapoptotic, antifibrotic, and anti-inflammatory effects[42-44]. Recently, data on ischemia-reperfusion rat model showed a T3 regulatory activity on hypertrophy through the regulation of receptors AT1R, AT2R, and MAS1 and, thus, on the cardiac renin-angiotensin-system (RAS). T3 administration to the ischemic heart revealed an anti-hypertrophic effect inducing the so-called “protective arm” (involving Ang1-7/AT2/MAS1) and inhibiting the “detrimental arm” (involving AngII/AT1R) of RAS[45].
Moreover, TH affect the microRNA cardiac profile and upregulate 19% of microRNAs inducing physiological myocardial changes and downregulate 56% of microRNAs regulating pathological changes[46]. In experimental model of myocardial infarction, administration of TH shortly after coronary ligation induces determinant changes aiming to ameliorate the OxS-mediated damage and contraction efficiency; a-MHC expression and SERCA2/phospholamban ratio increased, whereas b-MHC expression decreased[47]. Moreover, the expression of protein kinase C (PKC) -α (which reduces contractility) and -ε (with pro-hypertrophic function) were significantly diminished[48,49]. Thus, TH principal effects on infarcted myocardium is to contribute to the reversion of fetal-like to normal adult contractile phenotype, in the attempt of restoring as much as possible a correct cardiac performance[49].
Conclusion
In conclusion, the identification of TH-responsive molecular pathways involving ROS will help to obtain more information on molecular mechanisms regulating the interplay of TH with CVD with the aim to develop new efficient therapeutical strategies. However, because of the complexity of CVD, no complete information can be given by a single biomarker and it is crucial to individuate a panel of OxS-induced molecules, relevant for heart disease progression and under TH regulation. Moreover, it is important to consider that TH influence not only major CV changes but also many subtle events that may have a role in the cardiovascular risk. So far, available data have been collected mainly by limited randomized controlled studies or experimental investigations. It is desirable that the efforts of future research might be oriented towards the applicability of well-designed longitudinal clinical trials where T3, T4, or combination therapy is administered in LT3S patients with altered CV conditions, with the ultimate aim to define the most appropriate personalized schedule of treatment. In this scenario, another possible clinical application may imply the counteraction of OxS-induced CV injury through specific drugs or antioxidant supplementation (e.g., vitamin D, selenium) and/or life-style interventions (e.g., exercise or diet habit). This strategy will open further possibilities to be evaluated in longitudinal studies, which may include the combination of antioxidants supplementation and/or TH replacement therapies.
Declarations
Authors’ contributionsContributed to the conception and writing of the review: Sabatino L
Contributed to the reviewing and editing of the review: Ndreu R, Vassalle C
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 2011;301:H2181-90.
2. Ho E, Karimi Galougahi K, Liu CC, Bhindi R, Figtree GA. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol 2013;1:483-91.
3. Resch U, Helsel G, Tatzber F, Sinzinger H. Antioxidant status in thyroid dysfunction. Clin Chem Lab Med 2002;40:1132-4.
4. Mancini A, Di Segni C, Raimondo S, et al. Thyroid Hormones, Oxidative Stress, and Inflammation. Mediators Inflamm 2016;2016:6757154.
5. Gaggini M, Traghella I, Vassalle C. The Thyroid-Oxidative Stress Axis in Heart Failure. In: Iervasi G, Pingitore A, Gerdes A, Razvi S, editors. Thyroid and Heart. Cham: Springer International Publishing; 2020. pp. 171-86.
6. Mishra P, Samanta L. Oxidative stress and heart failure in altered thyroid States. ScientificWorldJournal 2012;2012:741861.
7. Stocker R, Keaney JF Jr. Role of oxidative modifications in atherosclerosis. Physiol Rev 2004;84:1381-478.
9. Thollon C, Iliou JP, Cambarrat C, Robin F, Vilaine JP. Nature of the cardiomyocyte injury induced by lipid hydroperoxides. Cardiovasc Res 1995;30:648-55.
10. Schulz E, Wenzel P, Münzel T, Daiber A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid Redox Signal 2014;20:308-24.
11. Daiber A, Di Lisa F, Oelze M, et al. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br J Pharmacol 2017;174:1670-89.
12. Hernanz R, Briones AM, Salaices M, Alonso MJ. New roles for old pathways? A circuitous relationship between reactive oxygen species and cyclo-oxygenase in hypertension. Clin Sci (Lond) 2014;126:111-21.
13. Dubois-Deruy E, Peugnet V, Turkieh A, Pinet F. Oxidative Stress in Cardiovascular Diseases. Antioxidants (Basel) 2020;9:864.
14. Wang Y, Branicky R, Noë A, Hekimi S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol 2018;217:1915-28.
15. Mastorci F, Sabatino L, Vassalle C, Pingitore A. Cardioprotection and Thyroid Hormones in the Clinical Setting of Heart Failure. Front Endocrinol (Lausanne) 2019;10:927.
17. Kushnir A, Marks AR. The ryanodine receptor in cardiac physiology and disease. Adv Pharmacol 2010;59:1-30.
18. Koss KL, Kranias EG. Phospholamban: a prominent regulator of myocardial contractility. Circ Res 1996;79:1059-63.
20. Morkin E, Flink I, Goldman S. Biochemical and physiologic effects of thyroid hormone on cardiac performance. Progress in Cardiovascular Diseases 1983;25:435-64.
21. Mai W, Janier MF, Allioli N, et al. Thyroid hormone receptor alpha is a molecular switch of cardiac function between fetal and postnatal life. Proc Natl Acad Sci U S A 2004;101:10332-7.
22. Davis PJ, Goglia F, Leonard JL. Nongenomic actions of thyroid hormone. Nat Rev Endocrinol 2016;12:111-21.
23. Wickenden AD, Kaprielian R, Parker TG, Jones OT, Backx PH. Effects of development and thyroid hormone on K+ currents and K+ channel gene expression in rat ventricle. J Physiol 1997;504:271-86.
24. Cernohorský J, Kolár F, Pelouch V, Korecky B, Vetter R. Thyroid control of sarcolemmal Na+/Ca2+ exchanger and SR Ca2+-ATPase in developing rat heart. Am J Physiol 1998;275:H264-73.
25. Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest 1996;98:2572-9.
26. Blankenberg S, Barbaux S, Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis 2003;170:191-203.
27. Herbert J, Bono F, Savi P. The mitogenic effect of H 2 O 2 for vascular smooth muscle cells is mediated by an increase of the affinity of basic fibroblast growth factor for its receptor. FEBS Letters 1996;395:43-7.
28. Delafontaine P, Ku L. Reactive oxygen species stimulate insulin-like growth factor I synthesis in vascular smooth muscle cells. Cardiovasc Res 1997;33:216-22.
29. Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med 2012;366:54-63.
30. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res 2012;110:159-73.
31. Mukherjee SB, Das M, Sudhandiran G, Shaha C. Increase in cytosolic Ca2+ levels through the activation of non-selective cation channels induced by oxidative stress causes mitochondrial depolarization leading to apoptosis-like death in Leishmania donovani promastigotes. J Biol Chem 2002;277:24717-27.
32. Sheeran FL, Pepe S. Mitochondrial Bioenergetics and Dysfunction in Failing Heart. Adv Exp Med Biol 2017;982:65-80.
33. Sheeran FL, Pepe S. Energy deficiency in the failing heart: linking increased reactive oxygen species and disruption of oxidative phosphorylation rate. Biochim Biophys Acta 2006;1757:543-52.
34. Pfeffer MA. Left ventricular remodeling after acute myocardial infarction. Annu Rev Med 1995;46:455-66.
36. Sabatino L. TH Treatment in Patients with Cardiac Disorders: General Aspects and Rationale. In: Iervasi G, Pingitore A, Gerdes A, Razvi S, editors. Thyroid and Heart. Cham: Springer International Publishing; 2020. pp. 373-80.
37. Swynghedauw B, Delcayre C, Samuel JL, Mebazaa A, Cohen-Solal A. Molecular mechanisms in evolutionary cardiology failure. Ann N Y Acad Sci 2010;1188:58-67.
39. Wang B, Liu S, Li L, et al. Non-thyroidal illness syndrome in patients with cardiovascular diseases: A systematic review and meta-analysis. Int J Cardiol 2017;226:1-10.
40. Pingitore A, Landi P, Taddei MC, Ripoli A, L’Abbate A, Iervasi G. Triiodothyronine levels for risk stratification of patients with chronic heart failure. Am J Med 2005;118:132-6.
41. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev 2002;23:38-89.
42. Ortiz VD, de Castro AL, Campos C, et al. Effects of thyroid hormones on aortic tissue after myocardial infarction in rats. Eur J Pharmacol 2016;791:788-93.
43. Yao J, Eghbali M. Decreased collagen gene expression and absence of fibrosis in thyroid hormone-induced myocardial hypertrophy. Response of cardiac fibroblasts to thyroid hormone in vitro. Circ Res 1992;71:831-9.
44. Kmiec Z, Myśliwska J, Rachón D, Kotlarz G, Sworczak K, Myśliwski A. Natural killer activity and thyroid hormone levels in young and elderly persons. Gerontology 2001;47:282-8.
45. Sabatino L, Balzan S, Kusmic C, Iervasi G. Modification of gene expression profiling related to renin-angiotensin system in an ischemia/reperfusion rat model after T3 infusion. Mol Cell Biochem 2018;449:277-83.
46. Janssen R, Zuidwijk MJ, Kuster DW, Muller A, Simonides WS. Thyroid Hormone-Regulated Cardiac microRNAs are Predicted to Suppress Pathological Hypertrophic Signaling. Front Endocrinol (Lausanne) 2014;5:171.
47. Danzi S, Ojamaa K, Klein I. Triiodothyronine-mediated myosin heavy chain gene transcription in the heart. Am J Physiol Heart Circ Physiol 2003;284:H2255-62.
48. Rybin V, Steinberg SF. Thyroid hormone represses protein kinase C isoform expression and activity in rat cardiac myocytes. Circ Res 1996;79:388-98.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sabatino L, Ndreu R, Vassalle C. Oxidative stress and heart disease: the thyroid hormone mediation. Vessel Plus 2021;5:3. http://dx.doi.org/10.20517/2574-1209.2020.62
AMA Style
Sabatino L, Ndreu R, Vassalle C. Oxidative stress and heart disease: the thyroid hormone mediation. Vessel Plus. 2021; 5: 3. http://dx.doi.org/10.20517/2574-1209.2020.62
Chicago/Turabian Style
Sabatino, Laura, Rudina Ndreu, Cristina Vassalle. 2021. "Oxidative stress and heart disease: the thyroid hormone mediation" Vessel Plus. 5: 3. http://dx.doi.org/10.20517/2574-1209.2020.62
ACS Style
Sabatino, L.; Ndreu R.; Vassalle C. Oxidative stress and heart disease: the thyroid hormone mediation. Vessel Plus. 2021, 5, 3. http://dx.doi.org/10.20517/2574-1209.2020.62
About This Article
Copyright

Data & Comments
Data



 Cite This Article 14 clicks
Cite This Article 14 clicks

 Like This Article 23
likes
Like This Article 23
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.