
The role of vascular endothelium and exosomes in human protozoan parasitic diseases

Abstract
The vascular endothelium is a vital component in maintaining the structure and function of blood vessels. The endothelial cells (ECs) mediate vital regulatory functions such as the proliferation of cells, permeability of various tissue membranes, and exchange of gases, thrombolysis, blood flow, and homeostasis. The vascular endothelium also regulates inflammation and immune cell trafficking, and ECs serve as a replicative niche for many bacterial, viral, and protozoan infectious diseases. Endothelial dysfunction can lead to vasodilation and pro-inflammation, which are the hallmarks of many severe diseases. Exosomes are nanoscale membrane-bound vesicles that emerge from cells and serve as important extracellular components, which facilitate communication between cells and maintain homeostasis during normal and pathophysiological states. Exosomes are also involved in gene transfer, inflammation and antigen presentation, and mediation of the immune response during pathogenic states. Protozoa are a diverse group of unicellular organisms that cause many infectious diseases in humans. In this regard, it is becoming increasingly evident that many protozoan parasites (such as Plasmodium, Trypanosoma, Leishmania, and Toxoplasma) utilize exosomes for the transfer of their virulence factors and effector molecules into the host cells, which manipulate the host gene expression, immune responses, and other biological activities to establish and modulate infection. In this review, we discuss the role of the vascular endothelium and exosomes in and their contribution to pathogenesis in malaria, African sleeping sickness, Chagas disease, and leishmaniasis and toxoplasmosis with an emphasis on their actions on the innate and adaptive immune mechanisms of resistance.
Keywords
Introduction
The vascular endothelium
The vascular endothelium (VE) is a large endocrine organ consisting of a single layer (one cell thick) of endothelial cells (ECs). The VE mediates regulatory functions in cell proliferation, angiogenesis, permeability, blood flow, thrombosis, thrombolysis, coagulation, homeostasis, and inflammation[1,2]. It also acts as the barrier between vascular and parenchymal compartments of all organs and regulates the exchange of gases, immune cell trafficking, metabolism, and the spread of infections. The entire circulatory system, from the heart to the smallest capillaries, is layered with ECs, which carry out unique functions such as fluid filtration, maintaining the tone of the blood vessel, platelet and leukocyte interactions, neutrophil recruitment, and hormone trafficking[3,4]. The VE plays a major role in leukocyte recruitment from the vessel lumen and transit into tissue parenchyma[5,6], and it is also the precursor for both hematopoietic and endothelial lineages[7]. The role of the VE is governed by the presence of many membrane-bound receptors for molecules such as proteins, lipid transporting particles hormones, and metabolites[8,9]. Recent studies have identified the immunological role of the VE from the secretion of cytokines and chemokines to the expression of adhesion molecules and antigen presentation[10-12].
The VE is also involved in bacterial, viral, and protozoan infectious disease processes. However, the interactions between the pathogen and the VE and how they both influence the disease outcome needs to be explored further. A clearer understanding of these relationships may help in identifying potential targets for therapeutic intervention(s) to prevent and/or reduce disease severity.
Role of the VE in innate immunity
The VE serves as the first line of defense against the physical stimuli and chemical agonists that are present in the bloodstream during infection and disease by activating the inflammatory response when receptors on the ECs, such as the toll-like receptors (TLRs) and NOD-like receptors (NLRs), recognize Pathogen associated molecular patterns (PAMPs), damage associated molecular patterns (DAMPs), and pro-inflammatory cytokines such as interleukin (IL)-1β or TNF-α[13,14]. These recognition signals regulate the expression of pro-inflammatory genes (IL-1, IL-6, TNF-α, and IFN-γ), leukocyte recruitment, phagocytosis, and a subsequent adaptive immune response[14]. During the early phase of an inflammatory response, ECs are stimulated to release nitric oxide (NO), prostacyclin-2 (prostaglandin I2 or PGI2), and endothelin-1 to increase vasodilation by relaxing the surrounding smooth muscle cells[13,15]. Capillary permeability is further facilitated by the removal of occludin between EC junctions and inflammatory mediators such as kinins, cytokines, histamine, arachidonic acid, and complement components produced during this onset of inflammation[14]. VE activation upregulates expression of adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin by ECs to aid chemokine-guided leukocyte rolling and extravasation through the vessel wall into the target site of the tissue[14].
Role of the VE in adaptive immunity
The adaptive immune response can be mediated by the VE in multiple ways under certain conditions. For example, thrombin-activated platelets as discussed above, upregulate CD40L similar to activated T cells and are therefore able to interact with CD40 on the EC surface to promote cytokine and chemokine secretion, adhesion molecule expression, tissue factor release, and leukocyte recruitment[16,17]. Furthermore, the upregulation of adhesion molecules and chemokines by activated ECs also serves to selectively recruit specific T cell types in circulation[18]. During this recruitment, antigen presentation may occur between the ECs and T cells mediated by major histocompatibility complex (MHC). It is known that ECs express MHC Class I on their surface, as well as MHC Class II in certain vasculatures of the body[14]. Due to this presence of MHC, ECs can act as antigen-presenting cells (APCs) to present antigen to effector or memory T cells[19]. Interaction with T cell co-stimulatory or co-inhibitory molecules expressed on ECs, such as CD80, CD86, LFA-3, ICOS-L, PDL-1, CD40, and CD134L, also mediates the T cell response[14].
Due to their ability to act as APCs, ECs may generate certain T cell subsets to induce either inflammation or immune tolerance[14]. For instance, it has been shown that ECs support T cell proliferation and increases in the number of suppressor Treg cells[20] and can stimulate the production of pro-inflammatory Th17 cells under inflammatory conditions[21]. ECs are also directly recognized by effector memory CD4+ T cells to stimulate IFN-γ production and subsequent CD4+ Th1 polarization[22]. This phenomenon has been further supported by a study involving activation of the ECs mediated by the C3a and C5a anaphylatoxins, which induce a Th1 phenotype, an increase in IFN-γ production, and activation of B lymphoblasts[23]. Taken together, the evidence suggests that VE plays a critical role in the initial onset of inflammation, innate immunity, and subsequent adaptive immunity in response to diverse stimuli present within the bloodstream.
Exosomes
Exosomes are membrane-bound extracellular vesicles between 40 and 100 nm in diameter that have been recognized as important players in cell-to-cell signaling[24]. They are secreted into the extracellular space by ECs and various other cells, such as platelets, immunocytes, and smooth muscle cells[25-27] and are present in almost all biological fluids[28]. Extracellular exchange of exosomes containing various bioactive molecules takes place continuously between organelles to foster communication during homeostasis and diseased states[29]. Exosome production begins with the internalization of the cellular membrane through endocytosis to form an endosome, followed by the invagination of the endosomal membrane, which matures into multivesicular bodies (MVBs). The MVBs can then either be degraded by internal lysosomes or are transported to the cell membrane to undergo transcytosis or fusion and release of the contained liberates intraluminal vesicles into the extracellular space, becoming exosomes[28,30]. Contents carried by exosomes consist of different proteins, lipids, metabolites, RNA, and DNA, which may be exchanged between the exosomes and their target cells.
Originally, exosomes were thought to be involved only in the process of excretion of unnecessary/unwanted proteins, and several studies have revealed that cellular stress triggers an enhanced release of exosomes[31-34]. However, further studies have suggested the participation of exosomes in cellular communication associated with many physiological and pathological states due to their ability to influence the phenotype of recipient cells[24,28]. Exosomes can target cells through specific receptor binding to activate cell-to-cell signaling pathways, which induce specific functions[35]. Information from an exosome may be transferred to the recipient cell either by interacting at the cell surface or by endosomal uptake[36]. Functions of released exosomes include horizontal gene transfer, inflammation, antigen presentation, tumor progression, and mediation of the immune response during pathogenic states[35,37].
Exosomes have been isolated from protozoa, bacteria, viruses, and fungi but each has distinct exosome profiles with varying compositions[38]. Protozoan parasites modulate host cells by producing exosomes containing virulence factors and effector molecules. In this way, parasites can manipulate host gene expression, immune responses, and other factors that favor parasite growth, survival, and pathogenesis[39]. Exosomes released during infection may also facilitate host immunity[38,40].
Role of exosomes in innate immunity
Exosomes are thought to play an important role in the host immune response to infection. Reciprocal cross-talk between platelets and neutrophils is enabled by neutrophil-derived exosome transfer of arachidonic acid to platelets, which is enzymatically converted into thromboxane A2 (TxA2). Release of TxA2 from platelets contributes to the upregulation of ICAM-1 on neutrophils, which binds to the EC surface and enables their extravasation to the site of infection to engage microbes[41].
Exosomes are directly involved in immune signaling in as much as they can cargo pro-inflammatory and anti-inflammatory cytokines to target cells[42-45] as well as stimulate the secretion of these cytokines from recipient cells[35,46,47]. For instance, a recent study documented the role of apoptotic exosome-like vesicles in promoting the synthesis of the pro-inflammatory IL-1β in macrophages, thereby contributing to their activation[48]. In contrast, exosomes have also been shown to promote the production of the anti-inflammatory transforming growth factor-β (TGF-β) in macrophages leading to the inhibition of the innate immune response[47,49]. Murine LPS-stimulated macrophages produce exosomes containing endoplasmic reticulum aminopeptidase 1, TNF-α, IFN-γ, and CCL3, which induces phagocytosis and nitric oxide synthesis in adjacent recipient macrophages[47,50], important cellular mediators in clearing the microbial infection. Additionally, epithelial cell uptake of exosomes secreted by LPS-induced dendritic cells (DCs) has been shown to stimulate their activation and subsequent release of cytokines and chemokines to further the innate response[51,52].
PAMPs are vital for recognition of pathogens and activation of immune cells and have been found inside exosomes secreted by both infected cells[53,54] and pathogens[55-57]. Interaction of PAMP-containing exosomes with the innate immune cells can induce inflammation. This has been observed in bacteria-infected macrophages, which release exosomes-contained pathogen antigens, which in turn promote maturation of DCs and secretion of pro-inflammatory cytokines[58].
Role of exosomes in adaptive immunity
Exosomes not only influence innate immune responses but can also play a marked role in adaptive immunity by promoting immune activation or suppression. B-lymphocytes infected with Epstein-Barr virus were the first immune cells discovered to release exosomes. These exosomes were shown to contain peptide-bound MHC Class I and II, B7 co-stimulatory, and ICAM-1 adhesion molecules that could induce a specific T cell response through the direct presentation of MHC Class II antigen[51]. Later, DCs were also found to release exosomes, possessing MHC Class I and II and T cell co-stimulatory molecules, leading to direct antigen presentation and CD4+ and CD8+ T cell activation. DC-derived exosomes can also mediate antigen presentation through bystander DCs, by either cross-dressing or internalization of the exosomes and subsequent peptide transfer to the recipient cell MHC molecules. It is known that immature DCs have decreased T cell activation ability due to the lack of co-stimulatory molecules but do produce more antigen-presenting exosomes than mature DCs. Therefore, the transfer of exosomes from antigen-containing donor DCs to recipient bystander DCs allows indirect antigen presentation and activation of T cells, especially from the immature DCs, which are unable to successfully present the antigen on their own[51,59].
Furthermore, the CD4+ and CD8+ T cells can also constitutively secrete exosomes containing TCR/CD3 complexes, which is enhanced upon TCR activation[60] and can be either immune-activating or immune-suppressing depending on the microenvironment. Activated T cells can transfer exosomes to induce activation of resting T cells and enhance adaptive immune responses. For instance, CD3+ T cells activated with IL-2 and anti-CD3-secreted exosomes promote the proliferation of CD8+ T cells as well as their subsequent cytokine secretion[61].
Exosomes can also promote an immunosuppressed environment under certain circumstances. Exosomes produced by immature DCs have been shown to induce T cell anergy/deletion as well as activate CD4+ regulatory T (Treg) cells[38]. Treg cells promote an immunosuppressive environment by secreting exosomes containing anti-inflammatory molecules that inhibit IFN-γ secretion and CD4+ Th1 cell proliferation, as well as signal other T cells to differentiate into Treg cells. It has also been found that CD4+ T cells and certain B cells whose exosomes contain FasL can induce apoptosis in recipient T cells. Furthermore, ECs transfer anti-inflammatory miRNA through exosomes to mediate T cell responses and prevent chronic inflammation[62].
Human protozoan parasitic diseases
In humans, protozoan parasitic infections represent a substantial threat causing more than one million deaths annually[63]. According to the World Health Organization (WHO), it is estimated that protozoan parasitic infections occur in billions of people worldwide and are associated with significant mortality and morbidity and negatively impact many countries economically (WHO.org and[64]). The three most important protozoan diseases in humans are malaria, leishmaniasis, and African trypanosomiasis that cause disability-adjusted life years in millions of patients (WHO.org 2008 and 2010).
Many protozoan infections cause non-self-limiting chronic infections and neglected diseases. There are 20 diseases that affect more than one billion people in almost 149 tropical and subtropical countries and are responsible for approximately 12% of the total global health burden, which are categorized as Neglected Tropical Diseases (NTDs)[65] (https://www.who.int/neglected_diseases/diseases/en/). Due to the disease burdens, limited available effective treatments, and lack of vaccines, the WHO has classified NTDs such as leishmaniasis, Chagas disease, and Human African trypanosomiasis under the specialized program “Innovative and Intensified Disease Management”[66]. While most of these infections occur in developing countries, it is evident that developed countries are also affected[67]. In addition, the emergence of anti-microbial resistant strains, toxicity, and low effectiveness of the currently available treatments pose a substantial problem[68]. Globalization and socioeconomic conditions also play a major role in the emergence and spreading of specific protozoan parasitic infections[69]. Furthermore, while many protozoan infections remain asymptomatic, they can lead to death, especially among children. It is important to note that, unlike bacterial and viral infections, many protozoan parasitic infections do not have readily available vaccines[70]. The lack of reliable drugs, difficulties in vector control, and limited knowledge about these infections are also major barriers to preventing, controlling, and treating these protozoan parasitic infections. Understanding the host immune response to protozoan parasites is very important to develop vaccines and new drugs with low toxicity and high efficacy.
The most severe protozoan infections, such as malaria, leishmaniasis, Chagas diseases, Human African trypanosomiasis (HAT), and toxoplasmosis are transmitted through blood, although Chagas disease and Toxoplasmosis can also be transmitted through the consumption of infected meat and liquid[71]. Hematophagous arthropod vectors serve as intermediate hosts and transmit the parasites (Leishmania, Trypanosomes, and Plasmodium) between successive vertebrate hosts. Thus, these bloodborne and vector-transmitted infections involve complex interactions between the parasite and insect and mammalian hosts.
Below, we discuss the role of VE and exosomes in malaria, leishmaniasis, toxoplasmosis, Chagas disease, and HAT, as well as their effects on the innate and adaptive immune responses in these infections. The roles of VE and exosomes in these infections are summarized in Tables 1 and 2, respectively.
Roles and mechanisms of vascular endothelium in malaria, leishmaniasis, toxoplasmosis, Chagas disease, and HAT
| Parasite name | Disease | Role of VE | Ref. |
|---|---|---|---|
| Plasmodium spp. | Malaria | Expresses receptors for Plasmodium antigens | [72,73] |
| Reservoir for epoxide contains lipid signaling molecules and helps in multiplication of parasites | [74] | ||
| Produces low molecular weight growth factors, which enhance the parasite proliferation | [75] | ||
| Leishmania spp. | Leishmaniasis | Reservoirs for intra- and extracellular parasites | [76] |
| Releases nitric oxide (NO) and limits the spread of the disease | [77] | ||
| Expresses ICAM-1 in skin lesions in cutaneous disease, which helps lymphocyte migration s to sites of inflammation | [78] | ||
| Increases expression of VCAM-1, VEGF-A and VEGF-R in the skin lesions in cutaneous disease | [79-81] | ||
| Splenic endothelial cells express Ntrk2, helps in the pathological remodeling of the spleen in visceral disease | [82] | ||
| Toxoplasma. | Toxoplasmosis | Serves as replicative niche and provides the entrance to CNS | [83] |
| T. gondii infection leads to activation of cerebral endothelial cells, facilitating the spreading of the disease | [84] | ||
| Trypanosoma spp. | Chagas disease | Key role in the dissemination of parasites to the other organs | [85,86] |
| Produces various inflammatory molecules leading to trans-endothelial migration | [85,87,88] | ||
| Releases vasoactive molecules such as endothelin-1 and pro-inflammatory cytokines IL-1β, iL-6, TNF-α, and thromboxane A2 leading to the production of iNOS | [89-92] | ||
| Produces endothelin-1 and IL-1β, activated ERK1/2 and NF-κB, resulting in the induction of Cyclin-D1 in uninfected cells | [93-95] | ||
| Trypanosoma spp. | Human African trypanosomiasis | Serves as replicative niche | [96,97] |
| Produces inflammatory cytokines such as TNF-α, IL-6, and IL-8 | [98,99] | ||
| Induces the production of ICAM-1, E-selectin, and VCAM-1 to facilitate parasite migration into the central nervous system (CNS) | [96,98,100] | ||
| Facilitates parasite transit across the endothelium of cerebral blood vessels by the production of laminin-8, calcium, and papain-like cysteine proteases | [97,101] |
Roles and mechanisms played by exosomes in malaria, leishmaniasis, toxoplasmosis, Chagas disease, and HAT
| Disease | Exosomal Factors | Cell origin | Mode of Action | Ref. |
|---|---|---|---|---|
| Malaria | Parasitic components (protein, lipid, RNA, DNA) | Infected reticulocytes | Induces antigen presentation and elicit a long-term antibody protective immune response, increase memory CD4+ and CD8+ T cells | [102,103] |
| Pathogen genes | Infected RBCs | Facilitates cell-to-cell communication between parasites, promote differentiation to sexual forms | [104] | |
| Leishmaniasis | Virulence factors and effector proteins | Parasite | Induces secretion of IL-8 over TNF-α in host macrophages | [105] |
| Alters the cytokine response of monocytes through upregulating IL-10 and inhibiting TNF-α production | [106] | |||
| Inhibits IL-12p70, TNF-α, and IL-10 cytokine functions in monocyte-derived DCs and prevent DC-induced naïve T cell differentiation into mature Th1 cells | [107] | |||
| GP63 | Parasite | Exacerbates lesions due to increased production of inflammatory cytokine IL-17α and over the induction of IL-4 and IL-10 | [108] | |
| Infected macrophages | Regulates PTPs and TFs in target macrophages | [109] | ||
| Antigenic proteins | Infected DCs | Cleaves Dicer1 in hepatocytes to block miRNA-122 production, causing a decreased serum cholesterol level | [110] | |
| Toxoplasmosis | Antigenic proteins | Infected DCs | Induces protective spleen-derived Th1 and humoral immune responses with high levels of IgA antibody | [111,112] |
| Exosome | Parasite | Modulates macrophage activation through increased production of IL-12, TNF-α, and IFN-γ and a decrease in IL-10 | [113] | |
| PAMPs | Parasite | Induces protective cellular and humoral immune responses | [53] | |
| miRNA | Parasite | Activates inflammatory responses in nearby macrophages in a TLR- and MyD88-dependent manner. Interact and modulate host cells through gene regulation | [114] | |
| Chagas disease | Exosome | Infected blood cells | Protects extracellular parasites from complement-mediated lysis by binding the C3 convertase on the parasite surface and inhibiting C3 cleavage | [115-117] |
| Virulence factors and soluble proteins | Parasite | Helps parasites to invade host cells through the expression of transforming growth factor-beta (TGF-β) | [118] | |
| Complement regulatory and inhibitory proteins | Parasite | Enhances cell invasion and parasite survival by invading the innate immune system | [119,120] | |
| Avoids the complement system and increase the invasion of host cells | [121,122] | |||
| HAT | Virulence factors and proteins (SRA) | Parasite | Activates the innate and acquired immune responses and induces rapid clearance of erythrocytes to cause anemia and tissue damage | [123,124] |
Malaria
Malaria is the most prevalent tropical disease, which annually infects 300-500 million individuals worldwide (CDC.org). It is estimated that 2-3 million people are at risk every year, most of whom are children who die from the infection without the proper treatment along with the development of other complications. Malaria is prevalent among 90 countries, which represent 40% of the world’s population[125]. Malaria is caused by an intracellular protozoan parasite of the genus Plasmodium, in which five species are known to infect humans: P. falciparum, P. malariae, P. ovale, P. vivax, and P. knowlesi[126,127]. P. falciparum causes the most severe clinical form of the infection leading to major morbidity and mortality. Female Anopheles mosquitoes transmit this disease to humans when releasing sporozoites while taking a blood meal. These circulate in the blood and invade and mature in hepatocytes. Hepatic forms are released and invade erythrocytes where merozoites further increase in number and are released and invade other red blood cells (CDC.org: https://www.cdc.gov/malaria/about/disease.html).
Role of the VE in malaria
The VE plays a major role in host-parasite interactions and the severity of the malarial disease. It has been shown that P. falciparum antigens are present on the surface of infected erythrocytes and bind to the receptors expressed on the VE[72]. After invasion, Plasmodium modulates endothelial function either by direct adhesion to the EC receptors or by releasing parasite products that can induce EC activation, leading to the disruption of the EC barrier[73]. It has also been shown that histones released from merozoites (HeH) stimulate the production of inflammatory mediators by primary human dermal microvascular endothelial cells, supporting the pathogenic role of both host- and pathogen-derived histones in P. falciparum caused malaria[128].
Malaria caused by P. falciparum is associated with the cytoadherence to endothelial cells through the parasite ligand P. falciparum erythrocyte membrane protein 1 (PfEMP1). P. falciparum-infected RBCs sequester in blood capillaries through several endothelial cell cytoadherence receptor molecules such as CXCL1, ICAM1, CD36, and VCAM1[129] and release exosome-like vesicles to directly communicate between the parasites[104]. These extracellular vesicles and the other abnormal accumulation of metabolites play a critical role in the damage of the blood-brain barrier (BBB) in determining the severity of cerebral malaria (CM) caused by P. falciparum. CM is accompanied by coma, seizures, and focal neurological deficits, which contribute to a mortality rate of 15%-20% despite therapy[130]. After establishing infection, Plasmodium export many proteins, including epoxide hydrolases into the erythrocyte, which results in the alteration of fatty acid composition, leading to perturbed vascular function and sequestration of the parasite in the VE[74]. Exportation of PfEMP1 mediates the adhesion of infected erythrocytes to VE and placental syncytioblasts[131]. In addition, a recent study suggests that brain ECs produce low molecular weight growth factors, which stimulate the growth of P. falciparum in vitro. These growth factors potentially enhance parasite proliferation in erythrocytes in the brain microvasculature[75].
Role of exosomes in malaria
Production of exosomes from infected-host cells and Plasmodium species during infection correlates with higher malarial disease severity[39,132]. Supporting this idea, a study of P. falciparum revealed that exosome-like particles released from infected RBCs facilitate cell-to-cell communication among parasites through gene delivery. The P. falciparum protein PfPTP2 has been identified as a critical player in this mechanism. This cellular communication pathway promotes the multiplication of sexual forms (gametocytes), which is a key process to maintain malaria infection and increase transmission probability[104]. Furthermore, it has been hypothesized that blocking the synthesis of these exosome-like vesicles as a therapeutic target may lead to decreased parasite transmissibility[104].
Exosome production may serve as a means of host protection during malaria infection. In a study of P. yoelii-infected mice, parasite protein-containing exosomes were released from reticulocytes, which could induce antigen presentation and elicit a long-term antibody protective immune response when administered as a vaccine in naive mice. The production of IgG antibodies and recognition of the parasite-infected RBCs in response to the vaccination with released exosomes reduces the parasite load and leads to increased survival as well as reticulocytosis[102]. Vaccination of mice in vivo, as well as in vitro in human spleen cells, with CpG adjuvanted P. yoelii-infected reticulocyte-derived exosomes (rexPy) induces a spleen-dependent memory response against the parasite infection. This memory response is associated with the activation of spleen cells through rexPy uptake, leading to changes in the distribution of T cell subsets and, more specifically, an increase in memory CD4+ and CD8+ T cells[103]. Due to the immunoregulatory action, Plasmodium exosome particles are viable candidates in the development of future malaria vaccines[39].
Leishmaniasis
Leishmaniases are a group of neglected tropical diseases caused by infection with parasites belonging to the genus Leishmania, which are transmitted by the bite of infected sand flies. It is estimated that one billion people are at risk of infection and about 1.7 million new cases of leishmaniasis occur each year in 102 countries (https://www.who.int/leishmaniasis/resources/who_wer9122/en/). Due to the lack of efficient treatment options or a vaccine, leishmaniasis has become the second largest cause of death among parasitic infections after malaria. Leishmaniasis consists of a spectrum of clinical syndromes and is dependent on the species of infecting parasite. There are three main forms of the disease: localized or disseminated skin lesions [cutaneous leishmaniasis (CL) or diffuse cutaneous leishmaniasis caused by L. major and L. tropica], mucocutaneous disease (mucocutaneous leishmaniasis caused by L. Mexicana, L. braziliensis, and L. amazonensis), and systemic disease [visceral leishmaniasis (VL) caused by L. donovani and L. infantum]. Infection is initiated when infected female phlebotomine sand flies take a blood meal, leading to inoculation with infective promastigotes. Promastigotes are phagocytized by macrophages and neutrophils, which transform into and multiply as intracellular amastigotes, which can metastasize to distant organs. The lifecycle is complete when the infected macrophages are ingested by uninfected sandflies, which transform and replicate as promastigotes in the insect gut (https://www.cdc.gov/parasites/leishmaniasis/biology.html).
Role of the VE in leishmaniasis
The VE is critical for the initiation of inflammatory processes and vascular remodeling, including angiogenesis and lymphangiogenesis, which occur in the inflammatory microenvironments of both VL and CL infections[133,134]. Intra- and extracellular parasites attached to the wall of dermal blood vessels and the capillary lumen lead to the development of secondary infections and the spread of the disease, especially in endemic areas[135]. During CL, it has been shown that endothelial cells release NO, which counteracts the recruitment of granulocytes and limits the spreading of infection[135]. In CL infections, the venous endothelium of skin lesions expresses ICAM-1, which helps in the migration of lymphocytes to the site of inflammation[78]. CL infection with L. major has been shown to increase the expression of VCAM-1, which mediates the adhesion of mononuclear cells to the endothelial cells[79]. Both human and animal leishmanial infections lead to increased levels of vascular endothelial growth factor-A (VEGF-A) and its receptor (VEGF-R) in the skin[80,81]. Recently, Weinkopff et al.[133] showed that CL infection by L. major induces VEGF-A in macrophages in an ARNT/HIF dependent manner, leading to the limitation of inflammation and lymphangiogenesis. The expansion of the lymphatic network promotes lesion resolution, and inhibition of this process enhances the lesion development. In the VL model, L. donovani-infected mice aberrantly express neurotrophic tyrosine kinase receptor type-2 (Ntrk2) on splenic endothelial cells, which plays a role in pathologic remodeling of the spleen[82].
Role of exosomes in leishmaniasis
The role of exosomes in Leishmania infection is well studied, revealing that they serve as a key mode of delivery of Leishmania virulence factors and effector proteins to host cells during infection[136]. Both pathogen and host-derived exosomes have been identified in this process. Leishmania-derived exosomes can transport virulence factors into the host macrophages and induce secretion of IL-8 instead of TNF-β[105]. Proteomic analysis has revealed that one such virulence factor contained in L. major exosomes is the metalloprotease glycoprotein GP63, which regulates protein tyrosine phosphatases (PTPs) and transcription factors (TFs), such as NF-κB, in target macrophages[108]. PTPs prevent macrophage activation by inhibiting the secretion of pro-inflammatory IFN-γ, IL-12, and NO[137,138], which are important in host control of parasite infection. These act to modulate the immune response, diminishing inflammation in favor of parasite growth and survival. Exosomes released from L. donovani-infected macrophages contain GP63, which proteolyzes Dicer1 in hepatocytes to block miRNA-122, production leading to disease progression[109]. Exosomes released from L. donovani promastigotes can effectively alter the cytokine response of monocytes through the upregulation of IL-10 and inhibition of TNF-α production. Similarly, it has been observed that monocyte-derived DCs that have been exposed to parasite exosomes have inhibited levels of IL-12p70, TNF-α, and IL-10. Exosome-exposed DCs cannot induce naïve T cell differentiation into mature Th1 cells[106]. In contrast, Schnitzer et al.[110] showed that that vaccination with L. major antigens present in DC-derived exosomes can induce immune-protection against the infection.
Leishmania is also able to modify the production and content of exosomes in response to environmental stress (heat shock and pH) that mimic infection. Silverman et al.[105] showed that vesicle release from parasites can be increased by three-fold in response to heat shock. Interestingly, Atayde et al.[107] showed that Leishmania release exosomes within the lumen of the sand-fly midgut, which are ejected in the egested inoculum during by the sand-fly bite. These exosomes lead to exacerbated lesions in L. major and L. infantum models, possibly due to increased production of inflammatory cytokine IL-17α, and overproduction of IL-4 and IL-10, which are both known to suppress the Th1 responses and play a role in disease susceptibility.
Toxoplasmosis
Toxoplasmosis is caused by the obligate intracellular protozoan parasite, Toxoplasma gondii, which infects healthy and immunocompromised individuals worldwide. It is estimated that more than 11% of the population in the US and 60% of the population throughout the world are infected[139] (https://www.cdc.gov/parasites/toxoplasmosis/epi.html). The transmission of toxoplasmosis mainly occurs through ingesting raw meat containing T. gondii cysts (foodborne) or water containing oocysts from feline feces (waterborne). The parasite can also be transmitted congenitally when a woman acquires the infection during pregnancy[139], or very rarely through transplantation of organs[140]. Although felines act as definitive hosts for T. gondii, it can infect almost all nucleated mammalian and avian cells. The life cycle of T. gondii mainly involves an asexual phase within nucleated cells and a sexual phase, which occurs in felines. Ingested oocysts released in feline feces serve as the infectious stage for humans. Toxoplasmosis can cause miscarriage, stillborn infants, or severe central nervous system (CNS) disease; in adults, it can lead to multi-organ involvement including encephalitis, retino-uveitis, and pulmonary disease, especially in immunosuppressed hosts cerebral toxoplasmosis.
Role of the VE in toxoplasmosis
It is believed that T. gondii can modulate the gene expression of brain ECs and promote dissemination through the BBB. T. gondii transforms into motile extracellular forms (tachyzoites) that use transcellular or paracellular migration to cross the BBB and infect host cells. In this context, the ECs serves as a replicative niche for the entry of T. gondii to the CNS[83]. The most common clinical manifestation of toxoplasmosis is retinal infection. Recently, Furtado et al.[141] demonstrated that tachyzoites can cross the retinal endothelium to establish infection and that blocking this entrance leads to reduced diseased burdens. It has also been observed that T. gondii infection leads to induction and activation of cerebral blood vessel ECs[84].
Role of exosomes in toxoplasmosis
T. gondii was the first non-viral protozoan pathogen for which exosomes were identified[38]. Although T. gondii exosomes are continuously released during infection by tachyzoites, the bulk of released molecules are non-exosome associated excretory/secretory antigens constitute during the acute phase of infection[114]. An earlier study conducted by Aline et al.[111] revealed exosomes are important in generating protective immunity against T. gondii infection; T. gondii-pulsed DCs can effectively induce a spleen-derived Th1 immune response that is protective against acute and chronic infection It has also been shown that exosomes secreted by SRDCs induce a protective humoral immune response against the infection in syngeneic and allogeneic mice associated with high levels of IgA antibodies[112]. Furthermore, vaccination of mice before pregnancy with exosomes secreted by T. gondii-pulsed DCs protects pups from congenital infection due to a robust T cell response[142].
Li et al.[113] characterized T. gondii-derived exosomes as ~50 nm in size and containing HSP70, CD63, and T. gondii surface marker P30. These exosomes were shown to modulate macrophage activation through increased production of IL-12, TNF-α, and IFN-γ and a decrease in IL-10. Mice immunized with these exosomes exhibited cellular and humoral immune responses and were protected against acute infection[39,113]. Li et al.[143] also reported that T. gondii exosomes activate JNK signaling to elicit this innate immune response. Macrophages infected with T. gondii release exosomes containing PAMPs, which activate inflammatory responses in adjacent macrophages in a TLR- and myeloid differentiation factor 88 (MyD88)-dependent manner[53]. Some T. gondii exosomes contain miRNA that may interact and modulate the host cells through gene regulation[114]. Taken together, T. gondii-derived exosomes displayed significant immunogenic properties that make them viable candidates for vaccine production.
Chagas disease
The hemoflagellate Trypanosoma cruzi is the etiologic agent of Chagas disease, also termed American trypanosomiasis. Chronic infection by this parasite is characterized by chronic myocarditis, cardiomyopathy, and vasculopathy, as well as mega-organ syndromes[144-147]. It is estimated that ~8 million people are currently infected worldwide and that 20%-30% of those individuals develop sequelae of chronic infection. Chagas disease is the leading cause of heart failure in Latin America[148-151]. The parasite exists in four morphological forms: epimastigotes, insect metacyclic trypomastigotes, human trypomastigotes, and intracellular amastigotes[151]. Insect stage trypomastigotes (also termed metacyclic trypomastigotes) are the infective stage of the parasite, which are present in the feces of hematophagous triatomine insects, which contaminate wounded skin or mucous membranes[148,152]. Metacyclic trypomastigotes invade nucleated host cells to establish the infection. Inside the host cells, they transform and replicate as intracellular amastigotes, eventually differentiating into blood-stage trypomastigotes and exit the host cells to disseminate to multiple organs, including the heart and gastrointestinal tract of the mammalian hosts[148].
Role of the VE in Chagas disease
T. cruzi infects various types of cells including cardiac myocytes, GI-tract, smooth muscle cells, and the VE[95,153]. The VE plays a key role in the dissemination of T. cruzi, as parasites engage ECs during the initial stages of infection[85,86]. As the infection progresses, VE release inflammatory molecules by direct physical disruption of ECs, and parasites undergo trans-endothelial migration with the help of the parasite protease, cruzipain[85,87,88]. Cruzipain cleaves the human kininogen into bradykinin, an inflammatory mediator of endothelial permeability[85,154,155]. Studies have shown that T. cruzi induces ECs release of vasoactive molecules endothelin-1, pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α), and thromboxane A2, which trigger the production of iNOS and nitrosative stress[89-92]. Moreover, increased levels of TNF-α exacerbate endothelial COX-2/TXA2/TP/superoxide signaling[92]. The infection of T. cruzi also activates the NF-κB that accumulates in the nucleus and activates many genes specific to endothelial pathophysiology[95].
In addition to the vascular damage, T. cruzi invades VE through the secretion of neuraminidase that removes sialic acid from the infected ECs[156,157]. Collectively, all of these factors compromise the activity of ECs and cause vasculopathy, as demonstrated by vasospasm, focal ischemia, reduction in the blood flow, increased platelet aggregation, and elevated levels of thromboxane A (2) and endothelin-1[89,158,159]. Studies have shown that T. cruzi-infected ECs secrete endothelin-1 and interleukin-1beta (IL-1β), which activate extracellular signal-regulated kinases 1 and 2 (ERK1/2) and NF-κB, resulting in the expression of cyclin-D1 in uninfected smooth muscle cells[93-95]. The activation of these pathways involved in the interaction between T. cruzi and the VE are likely to play a major role in the inflammatory responses after vascular injury and endothelial dysfunction and could be potential targets for therapies to control parasite dissemination[65,85].
It is estimated that 10%-30% of patients infected with T. cruzi progress to the chronic stage manifested by cardiomyopathy and prothrombotic/inflammatory status[160]. Pathophysiological mechanisms such as activation of the endothelium and microvascular alterations occur during the cardiac damage. It is known that thromboxane A2 increases platelet aggregation and that inhibiting the formation of thromboxane A2 by aspirin alters the course of Chagas disease in both acute and chronic phases[161]. Benznidazole, a widely used drug for the treatment of Chagas disease, also acts by preventing endothelial damage caused by T. cruzi[162]. Cholesterol-lowering drugs such as simvastatin have been shown to decrease the endothelial activation and, in combination with benznidazole, improve the pathophysiological condition of chronic Chagas disease patients[163]. Recent discoveries have provided insights into how T. cruzi escapes the BBB and rapidly migrates across the ECs without disrupting the integrity of the monolayer or altering the permeability. Coates et al.[85] identified that this process is facilitated by bradykinin and CCL2, which may be considered in the development of new therapeutic strategies for Chagas disease.
Role of exosomes in Chagas disease
Parasite and host cell exosomes play a role in the pathogenesis in Chagas disease. The release of an elevated number of exosomes is essential for host-parasite interaction, intercellular communication, and enhanced parasite survival[120]. The infection of T. cruzi induces blood cells to release exosomes through a Ca2+- dependent manner[164]. The released exosomes protect extracellular trypomastigotes from complement-mediated lysis by binding to C3 convertase on the T. cruzi surface and inhibiting C3 cleavage[115-117]. Exosomes also aid the parasites to invade the host cells through the expression of TGF-β. The communication between cells takes place through the release of exosomal contents including cytokines, peptides, hormones, microRNA, and numerous bioactive substances, which act as a function of innate immunity[165,166]. Exosomes released by T. cruzi promote cell invasion and parasite survival by modulating the innate immune system and producing several virulence factors including the glycoprotein 85 (gp85), trans-sialidase, phosphatase, and the soluble proteins[116,119,120,167]. Thus, the exosomes released by the host cells and parasites during infection play a vital role in the invasion of the innate immune system, parasite survival, and the establishment of infection in Chagas disease.
Human African trypanosomiasis
Human African trypanosomiasis (HAT) is caused by two subspecies of Trypanosoma brucei: T.b. gambiense that leads to the chronic form of HAT known as West African trypanosomiasis and T.b. rhodesiense that leads to the acute form of HAT known as East African trypanosomiasis[168-170]. A third subspecies of T. b. brucei infects cattle and very rarely infects the human host. HAT is transmitted to mammalian hosts by the bite of infected tsetse flies. During a blood meal, the metacyclic trypomastigotes are injected into the skin of the host, eventually entering the lymphatic and blood vessels. As parasites transform into blood trypomastigotes, they are disseminated throughout the body. The life cycle is completed when trypomastigotes infect feeding tsetse flies, wherein they transform and replicated into insect stage parasites[168,171]. Unlike the other protozoan parasites, the entire life cycle of African trypanosomes consists of extracellular stages, which alternately infect mammalian and insect hosts (CDC.gov, https://www.cdc.gov/parasites/sleepingsickness/biology.html). Although T. brucei infection occurs through the hemolymphatic stage in the initial systemic stage, the second phase is mainly characterized as a central nervous system disease, due to parasite invasion of brain tissue, leading to the altered sensorium, seizures, coma, and death. These symptoms are the reason that HAT is also referred to as African sleeping sickness.
Role of the VE in HAT
Bloodstream forms of T. brucei multiply to high density and eventually invade the central nervous system through the penetration of the VE. Although the mechanism through which the T. brucei cross the BBB are yet to be fully understood, it has been shown that T. brucei uses a multi-step process using the host derived factors including the cytokines IFNα/β, IFNγ, TNF, ICAM-1, and CXCL10[97,172]. During infection, the VE cells are activated by the translocation of NF-κB, due to action of parasite trans-sialidase, to the nucleus and the induction pro-inflammatory cytokines such as TNF-α, IL-6, and IL-8. This process plus the induction of other soluble factors such as the adhesion molecules (ICAM-1, E-selectin, and VCAM-1)[100] culminates in leukocyte recruitment and transmigration of trypanosomes from the VE to the CNS[96,98]. Studies have shown that T. brucei infection enhances the eNOS protein expression, and enhanced NO production leads to elevated vasodilation and vascular permeability facilitating parasite invasion into the surrounding tissues and the central nervous system[173].
Parasite phospholipase C, protein kinase, and the parasite cysteine protease brucipain also participate in transmigration of trypanosomes into the CNS[101,174]. Furthermore, it has been shown that T. brucei crosses the VE of cerebral blood vessels of mice through interaction with laminin 8 of the ECs[97]. The transmigration of T. brucei through the vascular endothelium also depends on the calcium and the papain-like cysteine proteases[101]. Collectively, this shows that interaction with the VE depends on various factors that are essential to penetrate the BBB and infect the CNS.
Role of exosomes in HAT
The progression of HAT is modulated by several factors including macrophage hyper-activation, uncontrolled production of TNF, and the transfer of virulence factors by exosomes[123,175]. Host-derived exosomes play a major role in host defense and are targeted as vaccine candidates, whereas parasite-derived exosomes transduce signal(s) to the host cells to establish infection[39,176,177]. A study has shown that a spliced ladder RNA (SL RNA) is present in the exosomes of T. brucei that is essential in these parasites for the formation of all mature mRNA. The cells secreting these SL RNA-containing exosomes affect the social motility of these parasites[178]. The bloodstream form of the parasite is responsible for anemia and tissue damage in the mammalian host[124,179]. This immunopathological outcome is due to several proteins released from the exosomes that lead to sequential activation of the innate and acquired immune responses[180]. The parasites secrete several molecules through exosomes to gain access to the host cells. Likewise, T. brucei exosomes contain 156 proteins from diverse functional classes[123]. One study shows that T. brucei exosomes fuse with mammalian erythrocytes and causes rapid clearance of erythrocytes and promotes anemia[123,124]. Thus, the exosomal components (proteases) of the trypanosome could be the promising targets to control sleeping sickness[181].
Concluding remarks
Here, we discuss the important roles played by VE and exosomes in some major protozoan parasitic diseases. Exosomes serve as a carrier of effector molecules that modulate the host immune response in establishing infection. The content of exosomes provides an effectual means to control the protein expression in both parasite and host cells. While parasite-derived exosomes play a key role in establishing infections through intercellular communication and signaling mechanisms, the host-derived exosomes also play a major role in the host-defense mechanism. Understanding the mechanism of the exosomal component on the host immune system in causing parasitic disease may help in the development of a novel approach of diagnostic tools and treatment. Further research on exosomes is necessary to search for the candidate vaccine and drug development.
The VE provides effective immunological homeostasis that controls the inflammatory response, mainly through the production of cytokines. The VE is an important target of parasite invasion and the parasite interaction on the VE is responsible for the development of clinical manifestations. The trans-endothelial migration of parasites is the major key step in establishing infection. Thus, more experimental studies are needed to provide insights on the interaction of blood parasites and VE, trans-endothelial migration, and the role of endothelial cytokine mediators in parasite dissemination. A better understanding may reveal the way to find more anti-parasitic regimens.
To summarize, both the VE and exosomes regulate the entry of parasites, their multiplication, signaling between the parasite and host cells, and dissemination to the other organs of the hosts. In addition, the VE and exosomes modulate both the innate and adaptive immune responses and maintain the integrity of the inflammatory process (summarized in Figure 1 and Tables 1 and 2). However, further studies are needed for a thorough understanding of the mechanisms and roles played by the VE and exosomes in parasite survival and disease progression. The novel mechanisms regulated by the VE and exosomes can be considered as potential therapeutic targets to treat and control these human protozoan diseases.
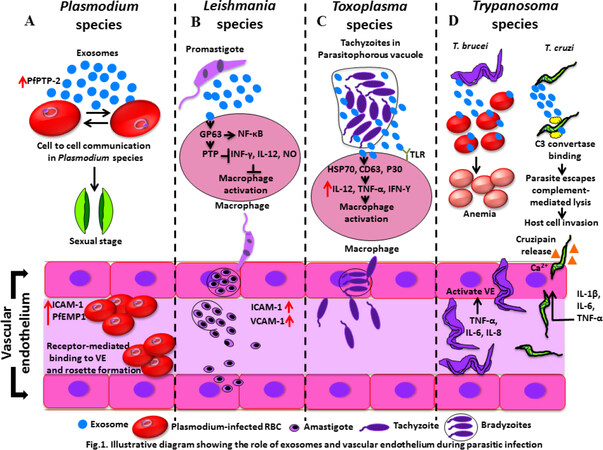
Figure 1. Schematic representation of cellular and molecular mechanisms played by vascular endothelium (VE) and exosomes in Plasmodium, Leishmania, Toxoplasma, and Trypanosoma spp. Infection. A: P. falciparum protein PfPTP-2 released through the exosomes from infected red blood cells (RBCs) facilitates cell-to-cell communication and promotes the differentiation of sexual forms of the parasites. P. falciparum erythrocyte membrane protein-1 (PfEMP1) and intercellular adhesion molecule-1 (ICAM-1) mediate the adhesion of infected erythrocytes to the VE and placental syncytioblasts; B: Leishmania parasites transport glycoproteins such as GP63 into the host cells through exosomes and regulate the protein tyrosine phosphatases (PTPs) and transcription factors such as NF-κB in macrophages. The PTPs prevent macrophage activation by inhibiting the secretion of IFN-γ, IL-12, and nitric oxide (NO). Leishmania infection also increases the expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) to initiate an inflammatory response after migrating the mononuclear cells and lymphocytes to the endothelial cells; C: T. gondii parasites release exosomes, which contain HSP70 and CD63, as well as the T. gondii surface marker P30. These exosomes induce the production of IL-12, TNF-α, and IFN-γ and modulate macrophage activation; D: T. brucei releases exosomes that are deposited and fused to RBCs. The virulence factors of exosomes result in RBC membrane alteration and anemia. In addition, T. brucei activates the vascular endothelial cells by producing TNF-α, IL-6, and IL-8. The exosomes of T. cruzi contain C3 convertase binding protein, which helps the parasites to escape the complement-mediated lysis. T. cruzi releases cruzipain and invades vascular endothelium through a Ca++-dependent mechanism
Declarations
Author’s contributionsContributed to the review conception, design, and writing of the manuscript: Varikuti S, Jha BK, Holcomb EA
Read and approved the final version of the manuscript: Varikuti S, Jha BK, Holcomb EA, McDaniel JC, Karpurapu M, Srivastava N, McGwire BS, Satoskar AR, Parinandi NL
Extended expertise in exosome biology: McDaniel JC
Offered expertise in exosome biology and macrophage biology: Karpurapu M, Srivastava N
Contributed expertise in infectious diseases and edited the manuscript: McGwire BS
Offered expertise in the protozoan parasitic diseases and immunology: Satoskar AR
Contributed expertise in vascular endothelial biology, signaling, and structure and function of exosomes and had the complete oversight of preparing this manuscript: Parinandi NL
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported partly by grants from National Institutes of Health (R01AG059981 and 5R21AI138555-02).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Valbuena G, Walker DH. The endothelium as a target for infections. Annu Rev Pathol 2006;1:171-98.
3. Durand MJ, Gutterman DD. Diversity in mechanisms of endothelium-dependent vasodilation in health and disease. Microcirculation 2013;20:239-47.
4. Verhamme P, Hoylaerts MF. The pivotal role of the endothelium in haemostasis and thrombosis. Acta Clin Belg 2006;61:213-9.
5. Luscinskas FW, Ma S, Nusrat A, Parkos CA, Shaw SK. Leukocyte transendothelial migration: a junctional affair. Semin Immunol 2002;14:105-13.
6. Biedermann BC. Vascular endothelium: checkpoint for inflammation and immunity. News Physiol Sci 2001;16:84-8.
7. Wang L, Li L, Shojaei F, Levac K, Cerdan C, et al. Endothelial and hematopoietic cell fate of human embryonic stem cells originates from primitive endothelium with hemangioblastic properties. Immunity 2004;21:31-41.
8. Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998;91:3527-61.
9. Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, et al. The vascular endothelium and human diseases. Int J Biol Sci 2013;9:1057-69.
10. Packer CS, Griffith SL, Roepke JE, Meiss RA, Rhoades RA. Myosin heavy chain isoform patterns do not correlate with force-velocity relationships in pulmonary arterial compared with systemic arterial smooth muscle. Adv Exp Med Biol 1991;304:397-402.
11. Zhu Y, Cui G, Miyauchi E, Nakanishi Y, Mukohira H, et al. Intestinal epithelial cell-derived IL-15 determines local maintenance and maturation of intra-epithelial lymphocytes in the intestine. Int Immunol 2020;32:307-19.
12. Emi-Sugie M, Shoda T, Futamura K, Takeda T, Ainai A, et al. Robust production of IL-33 and TSLP by lung endothelial cells in response to dsRNA stimulation. J Allergy Clin Immunol 2020; doi: 10.1016/j.jaci.2020.03.042.
13. Salvador B, Arranz A, Francisco S, Cordoba L, Punzon C, et al. Modulation of endothelial function by Toll like receptors. Pharmacol Res 2016;108:46-56.
14. Pons S, Arnaud M, Loiselle M, Arrii E, Azoulay E, et al. Immune consequences of endothelial cells’ activation and dysfunction during sepsis. Crit Care Clin 2020;36:401-13.
15. Quillon A, Fromy B, Debret R. Endothelium microenvironment sensing leading to nitric oxide mediated vasodilation: a review of nervous and biomechanical signals. Nitric Oxide 2015;45:20-6.
16. Danese S, Fiocchi C. Platelet activation and the CD40/CD40 ligand pathway: mechanisms and implications for human disease. Crit Rev Immunol 2005;25:103-21.
17. Joffre J, Hellman J, Ince C, Ait-Oufella H. Endothelial responses in sepsis. Am J Respir Crit Care Med 2020; doi: 10.1164/rccm.201910-1911TR.
18. Chimen M, Apta BH, McGettrick HM. Introduction: T cell trafficking in inflammation and immunity. Methods Mol Biol 2017;1591:73-84.
19. Savage CO, Brooks CJ, Harcourt GC, Picard JK, King W, et al. Human vascular endothelial cells process and present autoantigen to human T cell lines. Int Immunol 1995;7:471-9.
20. Lim WC, Olding M, Healy E, Millar TM. Human endothelial cells modulate CD4(+) T cell populations and enhance regulatory T cell suppressive capacity. Front Immunol 2018;9:565.
21. Taflin C, Favier B, Baudhuin J, Savenay A, Hemon P, et al. Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc Natl Acad Sci U S A 2011;108:2891-6.
22. Shiao SL, Kirkiles-Smith NC, Shepherd BR, McNiff JM, Carr EJ, et al. Human effector memory CD4+ T cells directly recognize allogeneic endothelial cells in vitro and in vivo. J Immunol 2007;179:4397-404.
23. Shivshankar P, Li YD, Mueller-Ortiz SL, Wetsel RA. In response to complement anaphylatoxin peptides C3a and C5a, human vascular endothelial cells migrate and mediate the activation of B-cells and polarization of T-cells. FASEB J 2020; doi: 10.1096/fj.201902397R.
24. Simons M, Raposo G. Exosomes--vesicular carriers for intercellular communication. Curr Opin Cell Biol 2009;21:575-81.
25. Liao J, Liu R, Yin L, Pu Y. Expression profiling of exosomal miRNAs derived from human esophageal cancer cells by Solexa high-throughput sequencing. Int J Mol Sci 2014;15:15530-51.
26. Saunderson SC, Dunn AC, Crocker PR, McLellan AD. CD169 mediates the capture of exosomes in spleen and lymph node. Blood 2014;123:208-16.
27. Wieckowski E, Whiteside TL. Human tumor-derived vs dendritic cell-derived exosomes have distinct biologic roles and molecular profiles. Immunol Res 2006;36:247-54.
28. Zhang J, Li S, Li L, Li M, Guo C, et al. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinformatics 2015;13:17-24.
29. Andersen JS, Mann M. Organellar proteomics: turning inventories into insights. EMBO Rep 2006;7:874-9.
30. Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci 2018;75:193-208.
31. Lehmann BD, Paine MS, Brooks AM, McCubrey JA, Renegar RH, et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res 2008;68:7864-71.
32. Beer L, Zimmermann M, Mitterbauer A, Ellinger A, Gruber F, et al. Analysis of the secretome of apoptotic peripheral blood mononuclear cells: impact of released proteins and exosomes for tissue regeneration. Sci Rep 2015;5:16662.
33. Xiao X, Yu S, Li S, Wu J, Ma R, et al. Exosomes: decreased sensitivity of lung cancer A549 cells to cisplatin. PLoS One 2014;9:e89534.
34. Kanemoto S, Nitani R, Murakami T, Kaneko M, Asada R, et al. Multivesicular body formation enhancement and exosome release during endoplasmic reticulum stress. Biochem Biophys Res Commun 2016;480:166-72.
35. Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol 2009;19:43-51.
36. Mathieu M, Martin-Jaular L, Lavieu G, Thery C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol 2019;21:9-17.
37. Schorey JS, Bhatnagar S. Exosome function: from tumor immunology to pathogen biology. Traffic 2008;9:871-81.
38. Schorey JS, Cheng Y, Singh PP, Smith VL. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep 2015;16:24-43.
39. Wu Z, Wang L, Li J, Wang L, Wu Z, et al. Extracellular vesicle-mediated communication within host-parasite interactions. Front Immunol 2018;9:3066.
40. Mantel PY, Marti M. The role of extracellular vesicles in Plasmodium and other protozoan parasites. Cell Microbiol 2014;16:344-54.
41. Rossaint J, Kuhne K, Skupski J, Van Aken H, Looney MR, et al. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat Commun 2016;7:13464.
42. Pizzirani C, Ferrari D, Chiozzi P, Adinolfi E, Sandona D, et al. Stimulation of P2 receptors causes release of IL-1beta-loaded microvesicles from human dendritic cells. Blood 2007;109:3856-64.
43. Gulinelli S, Salaro E, Vuerich M, Bozzato D, Pizzirani C, et al. IL-18 associates to microvesicles shed from human macrophages by a LPS/TLR-4 independent mechanism in response to P2X receptor stimulation. Eur J Immunol 2012;42:3334-45.
44. Cossetti C, Iraci N, Mercer TR, Leonardi T, Alpi E, et al. Extracellular vesicles from neural stem cells transfer IFN-gamma via Ifngr1 to activate Stat1 signaling in target cells. Mol Cell 2014;56:193-204.
45. Zhang HG, Liu C, Su K, Yu S, Zhang L, et al. A membrane form of TNF-alpha presented by exosomes delays T cell activation-induced cell death. J Immunol 2006;176:7385-93.
46. Perez PS, Romaniuk MA, Duette GA, Zhao Z, Huang Y, et al. Extracellular vesicles and chronic inflammation during HIV infection. J Extracell Vesicles 2019;8:1687275.
47. Chen Z, Larregina AT, Morelli AE. Impact of extracellular vesicles on innate immunity. Curr Opin Organ Transplant 2019;24:670-8.
48. Park SJ, Kim JM, Kim J, Hur J, Park S, et al. Molecular mechanisms of biogenesis of apoptotic exosome-like vesicles and their roles as damage-associated molecular patterns. Proc Natl Acad Sci U S A 2018;115:E11721-30.
49. Chen H, Kasagi S, Chia C, Zhang D, Tu E, et al. Extracellular vesicles from apoptotic cells promote tgfbeta production in macrophages and suppress experimental colitis. Sci Rep 2019;9:5875.
50. Goto Y, Ogawa Y, Tsumoto H, Miura Y, Nakamura TJ, et al. Contribution of the exosome-associated form of secreted endoplasmic reticulum aminopeptidase 1 to exosome-mediated macrophage activation. Biochim Biophys Acta Mol Cell Res 2018;1865:874-88.
51. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med 1996;183:1161-72.
52. Obregon C, Rothen-Rutishauser B, Gerber P, Gehr P, Nicod LP. Active uptake of dendritic cell-derived exovesicles by epithelial cells induces the release of inflammatory mediators through a TNF-alpha-mediated pathway. Am J Pathol 2009;175:696-705.
53. Bhatnagar S, Shinagawa K, Castellino FJ, Schorey JS. Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood 2007;110:3234-44.
54. Singh PP, LeMaire C, Tan JC, Zeng E, Schorey JS. Exosomes released from M. tuberculosis infected cells can suppress IFN-gamma mediated activation of naive macrophages. PLoS One 2011;6:e18564.
55. Kim MR, Hong SW, Choi EB, Lee WH, Kim YS, et al. Staphylococcus aureus-derived extracellular vesicles induce neutrophilic pulmonary inflammation via both Th1 and Th17 cell responses. Allergy 2012;67:1271-81.
56. Hong SW, Kim MR, Lee EY, Kim JH, Kim YS, et al. Extracellular vesicles derived from Staphylococcus aureus induce atopic dermatitis-like skin inflammation. Allergy 2011;66:351-9.
57. Prados-Rosales R, Baena A, Martinez LR, Luque-Garcia J, Kalscheuer R, et al. Mycobacteria release active membrane vesicles that modulate immune responses in a TLR2-dependent manner in mice. J Clin Invest 2011;121:1471-83.
58. Tan L, Wu H, Liu Y, Zhao M, Li D, et al. Recent advances of exosomes in immune modulation and autoimmune diseases. Autoimmunity 2016;49:357-65.
59. Zhang B, Yin Y, Lai RC, Lim SK. Immunotherapeutic potential of extracellular vesicles. Front Immunol 2014;5:518.
60. Blanchard N, Lankar D, Faure F, Regnault A, Dumont C, et al. TCR activation of human T cells induces the production of exosomes bearing the TCR/CD3/zeta complex. J Immunol 2002;168:3235-41.
61. Wahlgren J, Karlson Tde L, Glader P, Telemo E, Valadi H. Activated human T cells secrete exosomes that participate in IL-2 mediated immune response signaling. PLoS One 2012;7:e49723.
62. Anel A, Gallego-Lleyda A, de Miguel D, Naval J, Martinez-Lostao L. Role of exosomes in the regulation of T-cell mediated immune responses and in autoimmune disease. Cells 2019;8.
63. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380:2095-128.
64. Andrews KT, Fisher G, Skinner-Adams TS. Drug repurposing and human parasitic protozoan diseases. Int J Parasitol Drugs Drug Resist 2014;4:95-111.
65. Varikuti S, Jha BK, Volpedo G, Ryan NM, Halsey G, et al. Host-directed drug therapies for neglected tropical diseases caused by protozoan parasites. Front Microbiol 2018;9:2655.
66. Hotez PJ, Molyneux DH, Fenwick A, Kumaresan J, Sachs SE, et al. Control of neglected tropical diseases. N Engl J Med 2007;357:1018-27.
67. Fletcher SM, Stark D, Harkness J, Ellis J. Enteric protozoa in the developed world: a public health perspective. Clin Microbiol Rev 2012;25:420-49.
68. Cunha-Neto E, Chevillard C, Rodrigues MM, Bozza MT. Immunology and infection by protozoan parasites. Mediators Inflamm 2015;2015:504951.
69. Norman FF, Comeche B, Chamorro S, Perez-Molina JA, Lopez-Velez R. Update on the major imported protozoan infections in travelers and migrants. Future Microbiol 2020;15:213-25.
70. Burgess SL, Gilchrist CA, Lynn TC, Petri WA Jr. Parasitic protozoa and interactions with the host intestinal microbiota. Infect Immun 2017;85.
71. Chen MX, Ai L, Chen JH, Feng XY, Chen SH, et al. DNA microarray detection of 18 important human blood protozoan species. PLoS Negl Trop Dis 2016;10:e0005160.
72. Andersson AC, Resende M, Salanti A, Nielsen MA, Holst PJ. Novel adenovirus encoded virus-like particles displaying the placental malaria associated VAR2CSA antigen. Vaccine 2017;35:1140-7.
73. Gillrie MR, Ho M. Dynamic interactions of Plasmodium spp. with vascular endothelium. Tissue Barriers 2017;5:e1268667.
74. Spillman NJ, Dalmia VK, Goldberg DE. Exported epoxide hydrolases modulate erythrocyte vasoactive lipids during plasmodium falciparum infection. mBio 2016;7.
75. Khaw LT, Ball HJ, Mitchell AJ, Grau GE, Stocker R, et al. Brain endothelial cells increase the proliferation of Plasmodium falciparum through production of soluble factors. Exp Parasitol 2014;145:34-41.
76. Abedin TS, Thompson LK, Miller DO, Krupicka E. Structural and magnetic properties of a self-assembled spheroidal triakonta-hexanuclear Cu36 cluster. Chem Commun (Camb) 2003:708-9.
77. Fritzsche C, Schleicher U, Bogdan C. Endothelial nitric oxide synthase limits the inflammatory response in mouse cutaneous leishmaniasis. Immunobiology 2010;215:826-32.
78. ElHassan AM, Gaafar A, Theander TG. Antigen-presenting cells in human cutaneous leishmaniasis due to Leishmania major. Clin Exp Immunol 1995;99:445-53.
79. Henseleit U, Steinbrink K, Sunderkotter C, Goebeler M, Roth J, et al. Expression of murine VCAM-1 in vitro and in different models of inflammation in vivo: correlation with immigration of monocytes. Exp Dermatol 1995;4:249-56.
80. Weinkopff T, Konradt C, Christian DA, Discher DE, Hunter CA, et al. Leishmania major infection-induced VEGF-A/VEGFR-2 signaling promotes lymphangiogenesis that controls disease. J Immunol 2016;197:1823-31.
81. Araujo AP, Giorgio S. Immunohistochemical evidence of stress and inflammatory markers in mouse models of cutaneous leishmaniosis. Arch Dermatol Res 2015;307:671-82.
82. Dalton JE, Glover AC, Hoodless L, Lim EK, Beattie L, et al. The neurotrophic receptor Ntrk2 directs lymphoid tissue neovascularization during Leishmania donovani infection. PLoS Pathog 2015;11:e1004681.
83. Konradt C, Ueno N, Christian DA, Delong JH, Pritchard GH, et al. Endothelial cells are a replicative niche for entry of Toxoplasma gondii to the central nervous system. Nat Microbiol 2016;1:16001.
84. Deckert-Schluter M, Bluethmann H, Kaefer N, Rang A, Schluter D. Interferon-gamma receptor-mediated but not tumor necrosis factor receptor type 1- or type 2-mediated signaling is crucial for the activation of cerebral blood vessel endothelial cells and microglia in murine Toxoplasma encephalitis. Am J Pathol 1999;154:1549-61.
85. Coates BM, Sullivan DP, Makanji MY, Du NY, Olson CL, et al. Endothelial transmigration by Trypanosoma cruzi. PLoS One 2013;8:e81187.
86. Barrias ES, de Carvalho TM, De Souza W. Trypanosoma cruzi: entry into mammalian host cells and parasitophorous vacuole formation. Front Immunol 2013;4:186.
87. Todorov AG, Andrade D, Pesquero JB, Araujo Rde C, Bader M, et al. Trypanosoma cruzi induces edematogenic responses in mice and invades cardiomyocytes and endothelial cells in vitro by activating distinct kinin receptor (B1/B2) subtypes. FASEB J 2003;17:73-5.
88. Del Nery E, Juliano MA, Lima AP, Scharfstein J, Juliano L. Kininogenase activity by the major cysteinyl proteinase (cruzipain) from Trypanosoma cruzi. J Biol Chem 1997;272:25713-8.
89. Wittner M, Christ GJ, Huang H, Weiss LM, Hatcher VB, et al. Trypanosoma cruzi induces endothelin release from endothelial cells. J Infect Dis 1995;171:493-7.
90. Tanowitz HB, Gumprecht JP, Spurr D, Calderon TM, Ventura MC, et al. Cytokine gene expression of endothelial cells infected with Trypanosoma cruzi. J Infect Dis 1992;166:598-603.
91. Ashton AW, Mukherjee S, Nagajyothi FN, Huang H, Braunstein VL, et al. Thromboxane A2 is a key regulator of pathogenesis during Trypanosoma cruzi infection. J Exp Med 2007;204:929-40.
92. Silva JF, Capettini LS, da Silva JF, Sales-Junior P, Cruz JS, et al. Mechanisms of vascular dysfunction in acute phase of Trypanosoma cruzi infection in mice. Vascul Pharmacol 2016;82:73-81.
93. Mukherjee S, Huang H, Petkova SB, Albanese C, Pestell RG, et al. Trypanosoma cruzi infection activates extracellular signal-regulated kinase in cultured endothelial and smooth muscle cells. Infect Immun 2004;72:5274-82.
94. Huang H, Petkova SB, Cohen AW, Bouzahzah B, Chan J, et al. Activation of transcription factors AP-1 and NF-kappa B in murine Chagasic myocarditis. Infect Immun 2003;71:2859-67.
95. Huang H, Calderon TM, Berman JW, Braunstein VL, Weiss LM, et al. Infection of endothelial cells with Trypanosoma cruzi activates NF-kappaB and induces vascular adhesion molecule expression. Infect Immun 1999;67:5434-40.
96. Grab DJ, Kennedy PG. Traversal of human and animal trypanosomes across the blood-brain barrier. J Neurovirol 2008;14:344-51.
97. Masocha W, Robertson B, Rottenberg ME, Mhlanga J, Sorokin L, et al. Cerebral vessel laminins and IFN-gamma define Trypanosoma brucei brucei penetration of the blood-brain barrier. J Clin Invest 2004;114:689-94.
98. Girard M, Giraud S, Courtioux B, Jauberteau-Marchan MO, Bouteille B. Endothelial cell activation in the presence of African trypanosomes. Mol Biochem Parasitol 2005;139:41-9.
99. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 2009;1:a001651.
100. Ammar Z, Plazolles N, Baltz T, Coustou V. Identification of trans-sialidases as a common mediator of endothelial cell activation by African trypanosomes. PLoS Pathog 2013;9:e1003710.
101. Nikolskaia OV, de A Lima APC, Kim YV, Lonsdale-Eccles JD, Fukuma T, et al. Blood-brain barrier traversal by African trypanosomes requires calcium signaling induced by parasite cysteine protease. J Clin Invest 2006;116:2739-47.
102. Martin-Jaular L, Nakayasu ES, Ferrer M, Almeida IC, Del Portillo HA. Exosomes from Plasmodium yoelii-infected reticulocytes protect mice from lethal infections. PLoS One 2011;6:e26588.
103. Martin-Jaular L, de Menezes-Neto A, Monguio-Tortajada M, Elizalde-Torrent A, Diaz-Varela M, et al. Corrigendum: spleen-dependent immune protection elicited by CpG adjuvanted reticulocyte-derived exosomes from malaria infection is associated with changes in T cell subsets’ distribution. Front Cell Dev Biol 2016;4:153.
104. Regev-Rudzki N, Wilson DW, Carvalho TG, Sisquella X, Coleman BM, et al. Cell-cell communication between malaria-infected red blood cells via exosome-like vesicles. Cell 2013;153:1120-33.
105. Silverman JM, Clos J, de’Oliveira CC, Shirvani O, Fang Y, et al. An exosome-based secretion pathway is responsible for protein export from Leishmania and communication with macrophages. J Cell Sci 2010;123:842-52.
106. Silverman JM, Clos J, Horakova E, Wang AY, Wiesgigl M, et al. Leishmania exosomes modulate innate and adaptive immune responses through effects on monocytes and dendritic cells. J Immunol 2010;185:5011-22.
107. Atayde VD, Aslan H, Townsend S, Hassani K, Kamhawi S, et al. Exosome secretion by the parasitic protozoan leishmania within the sand fly midgut. Cell Rep 2015;13:957-67.
108. Hassani K, Shio MT, Martel C, Faubert D, Olivier M. Absence of metalloprotease GP63 alters the protein content of Leishmania exosomes. PLoS One 2014;9:e95007.
109. Ghosh J, Bose M, Roy S, Bhattacharyya SN. Leishmania donovani targets Dicer1 to downregulate miR-122, lower serum cholesterol, and facilitate murine liver infection. Cell Host Microbe 2013;13:277-88.
110. Schnitzer JK, Berzel S, Fajardo-Moser M, Remer KA, Moll H. Fragments of antigen-loaded dendritic cells (DC) and DC-derived exosomes induce protective immunity against Leishmania major. Vaccine 2010;28:5785-93.
111. Aline F, Bout D, Amigorena S, Roingeard P, Dimier-Poisson I. Toxoplasma gondii antigen-pulsed-dendritic cell-derived exosomes induce a protective immune response against T. gondii infection. Infect Immun 2004;72:4127-37.
112. Beauvillain C, Ruiz S, Guiton R, Bout D, Dimier-Poisson I. A vaccine based on exosomes secreted by a dendritic cell line confers protection against T. gondii infection in syngeneic and allogeneic mice. Microbes Infect 2007;9:1614-22.
113. Li Y, Liu Y, Xiu F, Wang J, Cong H, et al. Characterization of exosomes derived from Toxoplasma gondii and their functions in modulating immune responses. Int J Nanomedicine 2018;13:467-77.
114. Silva VO, Maia MM, Torrecilhas AC, Taniwaki NN, Namiyama GM, et al. Extracellular vesicles isolated from Toxoplasma gondii induce host immune response. Parasite Immunol 2018;40:e12571.
115. Cestari I, Ansa-Addo E, Deolindo P, Inal JM, Ramirez MI. Trypanosoma cruzi immune evasion mediated by host cell-derived microvesicles. J Immunol 2012;188:1942-52.
116. Cestari I, Ramirez MI. Inefficient complement system clearance of Trypanosoma cruzi metacyclic trypomastigotes enables resistant strains to invade eukaryotic cells. PLoS One 2010;5:e9721.
117. Wyllie MP, Ramirez MI. Microvesicles released during the interaction between Trypanosoma cruzi TcI and TcII strains and host blood cells inhibit complement system and increase the infectivity of metacyclic forms of host cells in a strain-independent process. Pathog Dis 2017;75.
118. Borges BC, Uehara IA, Dias LO, Brigido PC, da Silva CV, et al. Mechanisms of infectivity and evasion derived from microvesicles cargo produced by trypanosoma cruzi. Front Cell Infect Microbiol 2016;6:161.
119. Trocoli Torrecilhas AC, Tonelli RR, Pavanelli WR, da Silva JS, Schumacher RI, et al. Trypanosoma cruzi: parasite shed vesicles increase heart parasitism and generate an intense inflammatory response. Microbes Infect 2009;11:29-39.
120. Bayer-Santos E, Aguilar-Bonavides C, Rodrigues SP, Cordero EM, Marques AF, et al. Proteomic analysis of Trypanosoma cruzi secretome: characterization of two populations of extracellular vesicles and soluble proteins. J Proteome Res 2013;12:883-97.
121. Ramirez-Toloza G, Ferreira A. Trypanosoma cruzi evades the complement system as an efficient strategy to survive in the mammalian host: the specific roles of host/parasite molecules and trypanosoma cruzi calreticulin. Front Microbiol 2017;8:1667.
122. Karasu E, Eisenhardt SU, Harant J, Huber-Lang M. Extracellular vesicles: packages sent with complement. Front Immunol 2018;9:721.
123. Szempruch AJ, Sykes SE, Kieft R, Dennison L, Becker AC, et al. Extracellular vesicles from trypanosoma brucei mediate virulence factor transfer and cause host anemia. Cell 2016;164:246-57.
124. Stijlemans B, De Baetselier P, Magez S, Van Ginderachter JA, De Trez C. African trypanosomiasis-associated anemia: the contribution of the interplay between parasites and the mononuclear phagocyte system. Front Immunol 2018;9:218.
126. Miller LH, Ackerman HC, Su XZ, Wellems TE. Malaria biology and disease pathogenesis: insights for new treatments. Nat Med 2013;19:156-67.
127. Kavunga-Membo H, Ilombe G, Masumu J, Matangila J, Imponge J, et al. Molecular identification of Plasmodium species in symptomatic children of Democratic Republic of Congo. Malar J 2018;17:334.
128. Gillrie MR, Lee K, Gowda DC, Davis SP, Monestier M, et al. Plasmodium falciparum histones induce endothelial proinflammatory response and barrier dysfunction. Am J Pathol 2012;180:1028-39.
129. Newbold C, Craig A, Kyes S, Rowe A, Fernandez-Reyes D, et al. Cytoadherence, pathogenesis and the infected red cell surface in Plasmodium falciparum. Int J Parasitol 1999;29:927-37.
130. Idro R, Marsh K, John CC, Newton CR. Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr Res 2010;68:267-74.
131. Kats LM, Proellocks NI, Buckingham DW, Blanc L, Hale J, et al. Interactions between Plasmodium falciparum skeleton-binding protein 1 and the membrane skeleton of malaria-infected red blood cells. Biochim Biophys Acta 2015;1848:1619-28.
132. Combes V, Taylor TE, Juhan-Vague I, Mege JL, Mwenechanya J, et al. Circulating endothelial microparticles in malawian children with severe falciparum malaria complicated with coma. JAMA 2004;291:2542-4.
133. Weinkopff T, Roys H, Bowlin A, Scott P. Leishmania infection induces macrophage vascular endothelial growth factor A production in an ARNT/HIF-dependent manner. Infect Immun 2019;87.
134. Chowdhury KD, Sen G, Sarkar A, Biswas T. Role of endothelial dysfunction in modulating the plasma redox homeostasis in visceral leishmaniasis. Biochim Biophys Acta 2011;1810:652-65.
135. Lugo-Yarbuh A, Valera M, Alarcon M, Moreno E, Premoli-Percoco G, et al. Detection of Leishmania (Viannia) braziliensis in vascular endothelium lesions of patients with localized cutaneous leishmaniasis. Invest Clin 2003;44:61-76.
136. Silverman JM, Chan SK, Robinson DP, Dwyer DM, Nandan D, et al. Proteomic analysis of the secretome of Leishmania donovani. Genome Biol 2008;9:R35.
137. Heinonen KM, Dube N, Bourdeau A, Lapp WS, Tremblay ML. Protein tyrosine phosphatase 1B negatively regulates macrophage development through CSF-1 signaling. Proc Natl Acad Sci U S A 2006;103:2776-81.
138. Blanchette J, Racette N, Faure R, Siminovitch KA, Olivier M. Leishmania-induced increases in activation of macrophage SHP-1 tyrosine phosphatase are associated with impaired IFN-gamma-triggered JAK2 activation. Eur J Immunol 1999;29:3737-44.
139. Furtado JM, Smith JR, Belfort R Jr, Gattey D, Winthrop KL. Toxoplasmosis: a global threat. J Glob Infect Dis 2011;3:281-4.
140. Derouin F, Pelloux H, Parasitology ESGoC. Prevention of toxoplasmosis in transplant patients. Clin Microbiol Infect 2008;14:1089-101.
141. Furtado JM, Bharadwaj AS, Chipps TJ, Pan Y, Ashander LM, et al. Toxoplasma gondii tachyzoites cross retinal endothelium assisted by intercellular adhesion molecule-1 in vitro. Immunol Cell Biol 2012;90:912-5.
142. Beauvillain C, Juste MO, Dion S, Pierre J, Dimier-Poisson I. Exosomes are an effective vaccine against congenital toxoplasmosis in mice. Vaccine 2009;27:1750-7.
143. Li Y, Xiu F, Mou Z, Xue Z, Du H, et al. Exosomes derived from Toxoplasma gondii stimulate an inflammatory response through JNK signaling pathway. Nanomedicine (Lond) 2018;13:1157-68.
144. Tanowitz HB, Kirchhoff LV, Simon D, Morris SA, Weiss LM, et al. Chagas’ disease. Clin Microbiol Rev 1992;5:400-19.
145. Morris SA, Tanowitz HB, Wittner M, Bilezikian JP. Pathophysiological insights into the cardiomyopathy of Chagas’ disease. Circulation 1990;82:1900-9.
146. Torres SH, Finol HJ, Montes de Oca M, Vasquez F, Puigbo JJ, et al. Capillary damage in skeletal muscle in advanced Chagas’ disease patients. Parasitol Res 2004;93:364-8.
147. Rossi MA. Microvascular changes as a cause of chronic cardiomyopathy in Chagas’ disease. Am Heart J 1990;120:233-6.
148. Bern C, Kjos S, Yabsley MJ, Montgomery SP. Trypanosoma cruzi and Chagas’ disease in the United States. Clin Microbiol Rev 2011;24:655-81.
150. Nunes MC, Dones W, Morillo CA, Encina JJ, Ribeiro AL, et al. Chagas disease: an overview of clinical and epidemiological aspects. J Am Coll Cardiol 2013;62:767-76.
151. Coura JR, Dias JC. Epidemiology, control and surveillance of Chagas disease: 100 years after its discovery. Mem Inst Oswaldo Cruz 2009;104 Suppl 1:31-40.
152. Burleigh BA, Andrews NW. The mechanisms of Trypanosoma cruzi invasion of mammalian cells. Annu Rev Microbiol 1995;49:175-200.
153. Tanowitz HB, Burns ER, Sinha AK, Kahn NN, Morris SA, et al. Enhanced platelet adherence and aggregation in Chagas’ disease: a potential pathogenic mechanism for cardiomyopathy. Am J Trop Med Hyg 1990;43:274-81.
154. Scharfstein J, Schmitz V, Morandi V, Capella MM, Lima AP, et al. Host cell invasion by Trypanosoma cruzi is potentiated by activation of bradykinin B(2) receptors. J Exp Med 2000;192:1289-300.
155. Andrade D, Serra R, Svensjo E, Lima AP, Ramos ES Jr, et al. Trypanosoma cruzi invades host cells through the activation of endothelin and bradykinin receptors: a converging pathway leading to chagasic vasculopathy. Br J Pharmacol 2012;165:1333-47.
156. Nardy AF, Freire-de-Lima CG, Perez AR, Morrot A. Role of trypanosoma cruzi trans-sialidase on the escape from host immune surveillance. Front Microbiol 2016;7:348.
157. Libby P, Alroy J, Pereira ME. A neuraminidase from Trypanosoma cruzi removes sialic acid from the surface of mammalian myocardial and endothelial cells. J Clin Invest 1986;77:127-35.
158. Petkova SB, Huang H, Factor SM, Pestell RG, Bouzahzah B, et al. The role of endothelin in the pathogenesis of Chagas’ disease. Int J Parasitol 2001;31:499-511.
159. Petkova SB, Tanowitz HB, Magazine HI, Factor SM, Chan J, et al. Myocardial expression of endothelin-1 in murine Trypanosoma cruzi infection. Cardiovasc Pathol 2000;9:257-65.
160. Herrera RN, Diaz de Amaya EI, Perez Aguilar RC, Joo Turoni C, Maranon R, et al. Inflammatory and prothrombotic activation with conserved endothelial function in patients with chronic, asymptomatic Chagas disease. Clin Appl Thromb Hemost 2011;17:502-7.
161. Molina-Berrios A, Campos-Estrada C, Lapier M, Duaso J, Kemmerling U, et al. Protection of vascular endothelium by aspirin in a murine model of chronic Chagas’ disease. Parasitol Res 2013;112:2731-9.
162. Molina-Berrios A, Campos-Estrada C, Lapier M, Duaso J, Kemmerling U, et al. Benznidazole prevents endothelial damage in an experimental model of Chagas disease. Acta Trop 2013;127:6-13.
163. Gonzalez-Herrera F, Cramer A, Pimentel P, Castillo C, Liempi A, et al. Simvastatin attenuates endothelial activation through 15-Epi-Lipoxin A4 production in murine chronic chagas cardiomyopathy. Antimicrob Agents Chemother 2017;61.
164. de Pablos Torro LM, Retana Moreira L, Osuna A. Extracellular vesicles in chagas disease: a new passenger for an old disease. Front Microbiol 2018;9:1190.
165. Tkach M, Thery C. Communication by extracellular vesicles: where we are and where we need to go. Cell 2016;164:1226-32.
166. Penfornis P, Vallabhaneni KC, Whitt J, Pochampally R. Extracellular vesicles as carriers of microRNA, proteins and lipids in tumor microenvironment. Int J Cancer 2016;138:14-21.
167. Jazin EE, Bontempi EJ, Sanchez DO, Aslund L, Henriksson J, et al. Trypanosoma cruzi exoantigen is a member of a 160 kDa gene family. Parasitology 1995;110:61-9.
168. Franco JR, Simarro PP, Diarra A, Jannin JG. Epidemiology of human African trypanosomiasis. Clin Epidemiol 2014;6:257-75.
169. Morrison LJ. Parasite-driven pathogenesis in Trypanosoma brucei infections. Parasite Immunol 2011;33:448-55.
170. Sternberg JM, Maclean L. A spectrum of disease in human African trypanosomiasis: the host and parasite genetics of virulence. Parasitology 2010;137:2007-15.
171. Gull K. The cell biology of parasitism in Trypanosoma brucei: insights and drug targets from genomic approaches? Curr Pharm Des 2002;8:241-56.
172. Amin DN, Rottenberg ME, Thomsen AR, Mumba D, Fenger C, et al. Expression and role of CXCL10 during the encephalitic stage of experimental and clinical African trypanosomiasis. J Infect Dis 2009;200:1556-65.
173. Viswambharan H, Seebeck T, Yang Z. Enhanced endothelial nitric oxide-synthase activity in mice infected with Trypanosoma brucei. Int J Parasitol 2003;33:1099-104.
174. Rodgers J, Steiner I, Kennedy PGE. Generation of neuroinflammation in human African trypanosomiasis. Neurol Neuroimmunol Neuroinflamm 2019;6.
175. Magez S, Lucas R, Darji A, Songa EB, Hamers R, et al. Murine tumour necrosis factor plays a protective role during the initial phase of the experimental infection with Trypanosoma brucei brucei. Parasite Immunol 1993;15:635-41.
176. Devhare PB, Ray RB. A novel role of exosomes in the vaccination approach. Ann Transl Med 2017;5:23.
177. Coakley G, Maizels RM, Buck AH. Exosomes and other extracellular vesicles: the new communicators in parasite infections. Trends Parasitol 2015;31:477-89.
178. Eliaz D, Kannan S, Shaked H, Arvatz G, Tkacz ID, et al. Exosome secretion affects social motility in Trypanosoma brucei. PLoS Pathog 2017;13:e1006245.
179. Amole BO, Clarkson AB Jr, Shear HL. Pathogenesis of anemia in Trypanosoma brucei-infected mice. Infect Immun 1982;36:1060-8.
180. Paulnock DM, Freeman BE, Mansfield JM. Modulation of innate immunity by African trypanosomes. Parasitology 2010;137:2051-63.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Varikuti S, Kumar Jha B, Holcomb EA, McDaniel JC, Karpurapu M, Srivastava N, McGwire BS, Satoskar AR, Parinandi NL. The role of vascular endothelium and exosomes in human protozoan parasitic diseases. Vessel Plus 2020;4:28. http://dx.doi.org/10.20517/2574-1209.2020.27
AMA Style
Varikuti S, Kumar Jha B, Holcomb EA, McDaniel JC, Karpurapu M, Srivastava N, McGwire BS, Satoskar AR, Parinandi NL. The role of vascular endothelium and exosomes in human protozoan parasitic diseases. Vessel Plus. 2020; 4: 28. http://dx.doi.org/10.20517/2574-1209.2020.27
Chicago/Turabian Style
Varikuti, Sanjay, Bijay Kumar Jha, Erin A. Holcomb, Jodi C. McDaniel, Manjula Karpurapu, Nidhi Srivastava, Bradford S. McGwire, Abhay R. Satoskar, Narasimham L. Parinandi. 2020. "The role of vascular endothelium and exosomes in human protozoan parasitic diseases" Vessel Plus. 4: 28. http://dx.doi.org/10.20517/2574-1209.2020.27
ACS Style
Varikuti, S.; Kumar Jha B.; Holcomb EA.; McDaniel JC.; Karpurapu M.; Srivastava N.; McGwire BS.; Satoskar AR.; Parinandi NL. The role of vascular endothelium and exosomes in human protozoan parasitic diseases. Vessel Plus. 2020, 4, 28. http://dx.doi.org/10.20517/2574-1209.2020.27
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 2
likes
Like This Article 2
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.