
Atherogenesis, atherosclerosis and related diseases: unresolved issues
 0
0
Atherosclerosis is highly prevalent and affects most of the elderly population. It often develops in parallel with type 2 diabetes mellitus, metabolic syndrome, obesity and an excessive body mass, all of which worsen a patient’s comorbidities. The clustering of these pathologies, clinical manifestations of atherosclerosis and conventional cardiovascular risk factors is often observed in tandem in epidemiological and clinical studies. While isolated risk factors can account for risk variability, the combination of several risk factors translates to a highly unfavorable risk ratio. This concept of the accrual of several risk factors supports the association between atherosclerotic disease and cardiometabolic abnormalities. On the other hand, this concept also suggests that underlying genetic and metabolic mechanisms may either be similar or different, and can therefore be subdivided into common and disease-specific risk factors. Common risk factors may be partially explained by one’s genetic background, but the role of genetic factors in the clustering of risk factors in individuals remains unclear. Uncertainty over the common pathogenetic mechanisms, complexity of phenotypes, and the biases from lifestyle factors and therapeutic intervention give rise to the need for further discussion on unresolved issues in atherogenesis, atherosclerosis and related diseases.
Inspired by the opportunity to review and consolidate results from recent findings in the field of vascular remodeling, we have launched this special issue of “Vessel Plus”. We aimed to gather the latest research from both basic science and clinical investigations on the pathogenesis of atherosclerosis-related diseases and metabolic pathologies, and molecular pathways for further development of targeted therapy.
It is notable that atherosclerosis is the common denominator and characteristic of the above mentioned pathologies. Atherosclerosis is also either the cause of, or sequelae from these pathologies. Therefore, studies on the molecular and cellular mechanisms of atherogenesis are of utmost importance. Over the last few decades, progress in the reduction of cardiovascular mortality has been achieved mainly due to successful cardiovascular surgery and healthier lifestyles, which suggests that the battle against atherosclerotic disease has been won. However, the reduced role of cardiovascular disease in overall mortality is observed only in several developed countries. Globally however, atherosclerosis and related diseases still remain as one of the leading causes of mortality. It is true that patients have gained additional years of life from surgery and lipid-lowering medications, but did the underlying atherosclerosis resolve? Have the cellular and molecular mechanisms of atherogenesis been altered? Was there progress in the development of new strategies in anti-atherosclerotic treatment? No. Thus, it is time to discuss both the novel and debatable aspects of atherosclerosis and atherogenesis, with due consideration for the latest developments in molecular and cellular biology in vascular medicine.
Atherosclerosis can generally be described as an excessive fibro-fatty, proliferative and inflammatory response to arterial wall damage and involves several cell types such as monocyte-derived macrophages, smooth muscle cells, dendritic cells, lymphocytes and platelets[1,2]. It is well known that at the level of the arterial wall, the deposition of intracellular cholesterol and foam cell formation are the typical features of early atherosclerosis[3,4]. Current understanding of the development of early atherosclerotic lesions is shown in Figure 1. In brief, circulating lipoproteins - low density lipoprotein (LDL), the remnants of very low density lipoproteins (VLDL remnants) and lipoprotein(a) - serve as sources of lipids which can accumulate further in the arterial wall[4-8]. After penetrating the subendothelial intima, lipoproteins may induce lipid accumulation in cells as the initial and key step in the formation of atherosclerosis lesion[9,10]. However, LDL must first undergo proatherogenic modification, rendering it atherogenic i.e. capable of inducing intracellular lipid deposition[11-14] and this requires interactions between lipoproteins and connective tissue matrix components of the intima. In turn, this leads to retention of lipoproteins in the arterial intima, which increases the possibility of further interaction with intimal cells[15,16] to undergo complex cellular reactions such as the binding and internalization of modified lipoproteins, and abnormal intracellular processing, culminating in foam cell formation[4,10,17,18]. In parallel, intimal cells also exhibit a proliferative burst, increased synthesis of proteins and the components of connective tissue matrix, a pro-inflammatory response with the synthesis and secretion of cytokines, and the presentation of bound lipoproteins as autoantigens[4,19]. Thus, all the major characteristics of early atherosclerosis (lipidosis, fibrosis, proliferation and inflammation) are demonstrated at the cellular level at this stage of atherogenesis. In cases of relatively successful resolution of the cellular reaction to pathogenic lipoproteins, early lesions may either undergo spontaneous regression, or transform into stable atherosclerotic plaques. Such a result should be considered as compensatory arterial remodeling. On the other hand, an adverse outcome is characterized by a chronic inflammatory response, recruitment of immunocompetent cells from the circulation, and a vicious cycle of lipid accumulation in the arterial wall[20-22]. This unfavorable result will lead to growth of the atherosclerotic plaque, development of new lesions and plaque instability that will in turn, manifest as a clinical event.
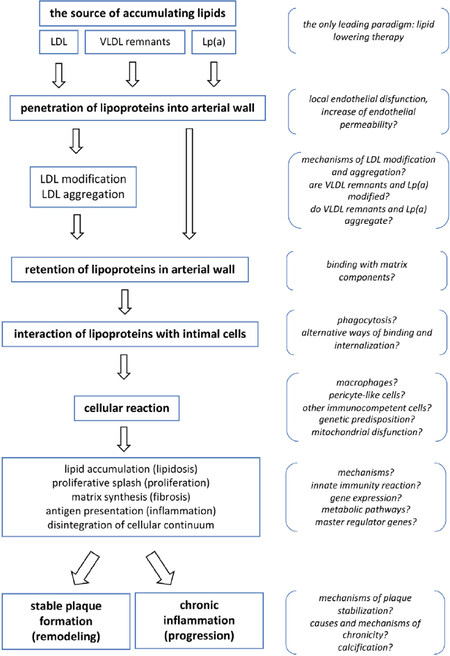
Figure 1. Current concepts of atherogenesis: schematic outline of key events and unresolved issues. LDL: low density lipoprotein; VLDL: very low density lipoprotein; Lp(a): lipoprotein (a)
Certainly, such schema is rather straightforward and oversimplified. It does not account for some mechanisms, such as the effectiveness of reverse cholesterol transport, high density lipoprotein (HDL) functioning, immunogenicity of modified LDL, the formation of atherogenic LDL-containing immune complexes, thrombotic events, etc., all of which have a role in atherogenesis. However, this scheme allows to demonstrate the unresolved issues with our knowledge on the molecular and cellular mechanisms of atherogenesis, i.e. those “white spots”, which obviously need further in-depth investigations.
The only existing paradigm in the prevention and treatment of atherosclerosis is extensive lipid lowering. This idea is based on the role of circulating lipoproteins as the source of cholesterol and as a key player in initializing atherogenesis. Thus, all current medical approaches are based on improving the lipid profile of blood plasma (lowering LDL cholesterol and triglycerides and increasing HDL cholesterol) and aims to eliminate major lipid risk factors for atherosclerosis. The development of alternative approaches for anti-atherosclerotic therapy through the targeting of other key pathogenetic mechanisms is extremely difficult due to the lack of fundamental knowledge on potential molecular and cellular targets for therapy and prevention. Among the latter are endothelial dysfunction and local violation of the permeability of the endothelial barrier, atherogenic modification of lipoproteins, retention of lipoproteins in the subendothelial intima of the arteries, alternative pathways of lipoprotein uptake by intimal cells, fibrotic and proliferative responses of intimal cells to modified lipoproteins, specificity of the reaction of different cells populating the intimal layer (smooth muscle cells, pericyte-like cells, macrophages, lymphocytes, other cells that migrated from circulation), the development of a local inflammatory reaction, ineffective resolution of local inflammation, local reaction of innate immunity, mitochondrial dysfunction, and the mechanisms for stabilization of atherosclerotic plaque and remodeling, etc.[Figure 1].
One of my own research interests is the mitochondrial genetics of atherosclerosis. There are several reasons for considering mutations occurring in mitochondrial DNA (mtDNA) as the mechanistic factor involved in atherogenesis. Atherosclerosis may be considered as an age-related degenerative pathology, accompanied by cell senescence which is generally characterized by reduced cell proliferation, irreversible growth arrest and apoptosis, epigenetic modifications, shortening of telomere length, increased mtDNA damage, and mitochondrial dysfunction[23]. The structural alterations of mitochondria and mtDNA damage are the most evident signs of mitochondrial aging[24]. Mutations of the mitochondrial genome can lead to structural defects in some energy-generating enzymes and transfer RNAs (tRNAs) synthesized directly in the mitochondria, thus playing a pathogenic role in the formation of atherosclerotic lesions. The decrease in concentration of these enzymes and tRNAs in mitochondria, and the resulting mitochondrial dysfunction contributes to oxidative stress, deterioration of ATP production and acceleration of atherogenesis[25].
Recently we have performed a series of studies on the relationships between (1) mtDNA variability and the changes in cellular composition of arterial atherosclerotic intima and the expression of apoptosis- and inflammation-related genes; (2) mtDNA variants, carotid atherosclerosis and conventional cardiovascular risk factors; and (3) individual mtDNA mutation burden and functional activity of cells in cell culture studies. The results of these studies strongly support the hypothesis on the atherogenic role of mtDNA mutations[26,27]. In further studies, we aimed to create cell models that reproduce the pathological cellular atherosclerotic phenotype with the use of promising approaches such as cytoplasmic hybrids (cybrids). At this stage of our studies, we have demonstrated that cybrid cells obtained from the homogeneous THP-1 line acquire completely different functional properties, and these changes were due solely to the functional activity of the donor mitochondria and the properties and mutational load of the introduced donor’s mtDNA[28,29]. However, the effects observed in cybrid cell lines are dependent not on some specific mtDNA variant, but on a unique combination of variants due to extremely high individual variation. Therefore, we needed more precise cellular models to investigate the intrinsic molecular mechanisms, which may be involved in the formation of the atherosclerotic phenotype due to mtDNA mutations; the use of direct mtDNA editing seemed plausible. We have launched a new research project, which would consistently carry out the design of liposomal delivery of nucleic acids and antisense RNA into cells and further into mitochondria, the delivery of CAS9 nuclease, sgRNA and ssODN recombination matrix, for the implementation of point mutations in mitochondrial genes[30]. This approach was approved and supported by the Russian Science Foundation. Currently, we have developed cationic liposomes with different addressable modules, evaluated the ability to deliver DNA and to transfect a vector expressing GFP into cells, developed the specific vector for introducing double-stranded breaks in mitochondrial genes, and for visualization of CAS9 localization in mitochondria[30]. These studies will help to evaluate the pathogenic role of deleterious mtDNA mutations in the formation of atherosclerotic phenotypes at the cellular level, and to find novel molecular targets for the prevention and treatment of atherosclerotic pathology.
In conclusion, I would like to thank the scientists and researchers who have contributed to this special issue of “Vessel Plus” and shared their own thoughts on recent fundamental, generalized and clinical findings, all of which are aimed at evaluating atherosclerosis-related and metabolic pathologies[31-34].
Declarations
Authors’ contributionsThe author contributed solely to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported in part by Russian Science Foundation, Grant No. 19-15-00297.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
3. Orekhov AN, Tertov VV, Novikov ID, Krushinsky AV, Andreeva ER, et al. Lipids in cells of atherosclerotic and uninvolved human aorta. I. Lipid composition of aortic tissue and enzyme-isolated and cultured cells. Exp Mol Pathol 1985;42:117-37.
4. Orekhov AN. LDL and foam cell formation as the basis of atherogenesis. Curr Opin Lipidol 2018;29:279-84.
5. Ellulu MS, Patimah I, Khaza’ai H, Rahmat A, Abed Y, et al. Atherosclerotic cardiovascular disease: a review of initiators and protective factors. Inflammopharmacology 2016;24:1-10.
6. Morita SY. Metabolism and modification of apolipoprotein B-containing lipoproteins involved in dyslipidemia and atherosclerosis. Biol Pharm Bull 2016;39:1-24.
7. Arnao V, Tuttolomondo A, Daidone M, Pinto A. Lipoproteins in atherosclerosis process. Curr Med Chem 2019;26:1525-43.
8. Nakajima K, Tanaka A. Atherogenic postprandial remnant lipoproteins; VLDL remnants as a causal factor in atherosclerosis. Clin Chim Acta 2018;478:200-15.
9. Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol 2019;16:727-44.
10. Maguire EM, Pearce SWA, Xiao Q. Foam cell formation: a new target for fighting atherosclerosis and cardiovascular disease. Vascul Pharmacol 2019;112:54-71.
11. Alipov VI, Sukhorukov VN, Karagodin VP, Grechko AV, Orekhov AN. Chemical composition of circulating native and desialylated low density lipoprotein: what is the difference? Vessel Plus 2017;1:107-15.
12. Summerhill VI, Grechko AV, Yet SF, Sobenin IA, Orekhov AN. The atherogenic role of circulating modified lipids in atherosclerosis. Int J Mol Sci 2019;20:E3561.
13. Orekhov AN, Sobenin IA. Modified and dysfunctional lipoproteins in atherosclerosis: effectors or biomarkers? Curr Med Chem 2019;26:1512-24.
14. Orekhov AN, Sobenin IA. Modified lipoproteins as biomarkers of atherosclerosis. Front Biosci (Landmark Ed) 2018;23:1422-44.
15. Borén J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol 2016;27:473-83.
16. Hurt-Camejo E, Camejo G. ApoB-100 lipoprotein complex formation with intima proteoglycans as a cause of atherosclerosis and its possible ex vivo evaluation as a disease biomarker. J Cardiovasc Dev Dis 2018;5:E36.
17. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med (Berl) 2017;95:1153-65.
18. Bäck M, Yurdagul A Jr, Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 2019;16:389-406.
19. Poznyak AV, Wu WK, Melnichenko AA, Wetzker R, Sukhorukov V, et al. Signaling pathways and key genes involved in regulation of foam cell formation in atherosclerosis. Cells 2020;9:E584.
20. Albany CJ, Trevelin SC, Giganti G, Lombardi G, Scottà C. Getting to the heart of the matter: the role of regulatory T-cells (Tregs) in cardiovascular disease (CVD) and atherosclerosis. Front Immunol 2019;10:2795.
21. Nasser MI, Zhu S, Huang H, Zhao M, Wang B, et al. Macrophages: first guards in the prevention of cardiovascular diseases. Life Sci 2020;250:117559.
22. Shi X, Gao J, Lv Q, Cai H, Wang F, et al. Calcification in atherosclerotic plaque vulnerability: friend or foe? Front Physiol 2020;11:56.
23. Wang C, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res 2012;111:245-59.
24. Yao YG, Kajigaya S, Feng X, Samsel L, McCoy JP Jr, et al. Accumulation of mtDNA variations in human single CD34+ cells from maternally related individuals: effects of aging and family genetic background. Stem Cell Res 2013;10:361-70.
25. Sobenin IA, Chistiakov DA, Bobryshev YV, Postnov AY, Orekhov AN. Mitochondrial mutations in atherosclerosis: new solutions in research and possible clinical applications. Curr Pharm Des 2013;19:5942-53.
26. Sobenin IA. Mitochondrial DNA damage in atherosclerosis. In: Parine NR, editor. Genetic Polymorphisms. Croatia: InTech; 2017. pp. 139-58.
27. Volobueva A, Grechko A, Yet SF, Sobenin I, Orekhov A. Changes in mitochondrial genome associated with predisposition to atherosclerosis and related disease. Biomolecules 2019;9:377.
28. Sazonova MA, Ryzhkova AI, Sinyov VV, Sazonova MD, Khasanova ZB, et al. Creation of cultures containing mutations linked with cardiovascular diseases using transfection and genome editing. Curr Pharm Des 2019;25:693-9.
29. Sazonova MA, Sinyov VV, Ryzhkova AI, Sazonova MD, Khasanova ZB, et al. Creation of cybrid cultures containing mtDNA mutations m.12315G>A and m.1555G>A, associated with atherosclerosis. Biomolecules 2019;9:499.
30. Sobenin IA, Sukhorukov VN, Sinyov VV, Khasanova ZB, Kalmykov VA, et al. Editing of mitochondrial genome: cationic liposomes as the perspective mean of targeted delivery of nucleic acids into mitochondria. Arch World Mitochondria Soc 2019;5:79.
31. Nemtsova V, Bilovol O, Ilchenko I, Shalimova A. Age-associated features of oxidative stress as marker of vascular aging in comorbid course of hypertension and type 2 diabetes mellitus. Vessel Plus 2018;2:27.
32. Archakova T, Nedosugova L. Risk factors for atherosclerosis and vascular calcification in patients with type 2 diabetes on long-term hemodialysis. Vessel Plus 2018;2:34.
33. Sazonova MA, Sinyov VV, Ryzhkova AI, Sazonova MD, Khasanova ZB, et al. MtDNA mutations linked with left ventricular hypertrophy. Vessel Plus 2019;3:5.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sobenin IA. Atherogenesis, atherosclerosis and related diseases: unresolved issues. Vessel Plus 2020;4:18. http://dx.doi.org/10.20517/2574-1209.2020.09
AMA Style
Sobenin IA. Atherogenesis, atherosclerosis and related diseases: unresolved issues. Vessel Plus. 2020; 4: 18. http://dx.doi.org/10.20517/2574-1209.2020.09
Chicago/Turabian Style
Sobenin, Igor A.. 2020. "Atherogenesis, atherosclerosis and related diseases: unresolved issues" Vessel Plus. 4: 18. http://dx.doi.org/10.20517/2574-1209.2020.09
ACS Style
Sobenin, IA. Atherogenesis, atherosclerosis and related diseases: unresolved issues. Vessel Plus. 2020, 4, 18. http://dx.doi.org/10.20517/2574-1209.2020.09
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 17 clicks
Cite This Article 17 clicks

 Like This Article 9
likes
Like This Article 9
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.