
Endothelial cell-derived extracellular vesicles in atherosclerosis: the emerging value for diagnosis, risk stratification and prognostication

Abstract
Endothelial cell-derived extracellular vesicles are produced by both activated and apoptotic endothelial cells, and play a pivotal role in various physiological conditions such as inflammation, repair, programmed cell death, and immune responses. There is a large body of evidence on the dysregulation of synthesis and secretion of several types of endothelial cell-derived extracellular vesicles, which can then trigger microvascular inflammation, atherosclerotic plaque formation, plaque rupture, thrombosis and endothelial dysfunction. The development of atherosclerosis and cardiovascular events is associated with an increased number of apoptotic, endothelial cell-derived vesicles and a decrease in activated, endothelial cell-derived vesicles. This review depicts the role of endothelial cell-derived extracellular vesicles in the manifestation and progression of atherosclerosis. We also discuss the clinical use and benefits of altering the immune phenotypes of extracellular vesicles originating from endothelial cells, to function as predictive biomarkers in both asymptomatic and subclinical atherosclerosis.
Keywords
Introduction
Atherosclerosis remains a leading cause of major cardiovascular events (MACEs) and cardiovascular (CV) diseases worldwide. It represents a serious economic burden on the healthcare system and is associated with high rates of mortality and morbidity[1]. While there has been a steady trend towards decreasing CV mortality from conditions associated with atherosclerosis such as stroke and myocardial infarction in developed countries over the last decade, mortality trends from coronary artery disease (CAD) have been more varied[2,3]. The decline in risk of potentially fatal complications from atherosclerosis in high-income countries has been reported to be closely connected with improved control of conventional CV risk factors such as hypertension, obesity and smoking, after implementation of current clinical guidelines[4,5]. Overall, the impact of atherosclerosis on MACEs, CV disease manifestation and disability in both developed and developing countries continues to be under investigation because full control of traditional CV risk factors (i.e., dyslipidemia, and diabetes mellitus) did not achieve disease reversal.
Non-adaptive remodeling in atherosclerosis leads to plaque formation, which is a consequence of events including endothelial dysfunction, impaired vascular repair, systemic and microvascular inflammation, and the migration, proliferation and phenotypic switch of smooth muscle cells[6]. There are numerous cellular and molecular mechanisms that contribute to the initiation and progression of vascular lesions in atherosclerosis including the infiltration of oxidized lipids into the sub-intima, transformation of macrophages from an anti-inflammatory into a pro-inflammatory phenotype, modification of the extracellular matrix due to imbalance between activities of matrix metalloproteinases and their inhibitors, the development of foam cells, over-production of inflammatory cytokines in the atherosclerotic plaque and within the sub-intima layer, expansion of the lipid core in the plaque and vascular tone dysregulation[7-9]. In turn, endothelial and vascular integrity are ensured by interaction of genetic and epigenetic programs that play a pivotal role in the maintenance of vascular homeostasis[10,11].
Previous pre-clinical and clinical studies have shown that extracellular vesicles (EVs) originate from progenitor and mature endothelial cells. EVs may act both locally and remotely as powerful regulators of vascular function and integrity through the transfer of biological information[12-14]. EVs are also involved in several pathological processes underlying progression of atherosclerosis such as systemic and microvascular inflammation, immunity, signal transduction, cell proliferation, differentiation, survival and apoptosis, as well as neovascularization, angiogenesis, thrombosis, and autophagy[14-16]. The purpose of this review is to summarize current knowledge on the role of endothelial cell-derived EVs in the manifestation and progression of atherosclerosis, and to discuss the clinical use and benefits of using altered immune phenotypes of these endothelial-cell derived EVs as predictive biomarkers in both asymptomatic and subclinical atherosclerosis.
Definition and nomenclature of EVs
EVs are a heterogenic population of secreted, membrane-enclosed particles. This includes exosomes, ectosomes, microvesicles, smal size microvesicles, micro particles, nano particles, apoptotic bodies and other subsets. Some (ectosomes and micro particles) are not distinct from each other, and several classification approaches (sedimentation speed-derived criteria, immune phenotype, origin, mechanism of release, and size) were applied to EV subsets to qualify them in some categories.
According to the last update of the Executive Committee of the International Society for EVs, EVs are defined as a mixture of particles ranging from 30-2000 nm in diameter, released by various types of viable cells through several mechanisms (blebbing and budding of endosomal or plasma membranes) and include exosomes, microvesicles and apoptotic bodies[17]. EV subtypes are defined according to numerous physical characteristics however, such as size (small, medium and large EVs with diameters < 100 nm, 100-200 nm and > 200 nm), density (low, middle, and high, with each range defined), biochemical composition (CD63+/CD81+, Annexin A5-stained), and descriptions of conditions or cell of origin (e.g., podocyte EVs, hypoxic EVs, large oncosomes, apoptotic bodies). Although the terms “exosome” and “microvesicle” are historically burdened by both manifold and inaccurate definitions, Table 1 reports both under the nomenclatures of EVs to easily understand the basic characterictics of several subtypes of EVs.
Nomenclature and basic characteristics of several subtypes of EVs
| Characteristics of EVs | Subpopulations of EVs | Ref. | ||
|---|---|---|---|---|
| Exosomes (small EVs) | Microvesicles (ectosomes, medium/large EVs) | Apoptotic bodies | ||
| Diameter, nm | 40-100 | 100-1000 | 50-2000 | [17] |
| Origin | Endocytic membrane | Cell membrane | Apoptotic cells | [18] |
| Mechanism of delivery | Ceramide-dependent, tetraspanin-dependent, and ESCRT-dependent exocytosis of multi vesicular bodies | Ca2+ depending phospholipid redistribution and Rho-kinase-mediated myosin light chain phosphorylation, facilitating budding and blebbing | thin membrane protrusion and blebbing of the apoptotic cells’ surface | [20,21] |
| Phosphatidylserine composition | Low | High | High | [22] |
| Complexity/granularity | High | High | Low | [26,27] |
| Components | Cytoplasmic and membrane molecules, proteins and lipids, tetraspanin’s receptors | Adhesive molecules (ICAMs, PECAM-1, MCAM), membrane regulatory proteins (Rab), lipids (SpL, PL, LPS, LPS) and receptors (tetraspanin’s receptors, LAIR-1, EGFR), enzymes (Rab GTPase, ERK, MLCK, TPI-1, HMGCL), immune system proteins (CD14, CD276, MiC-11), apoAII, SOD, β-actin, α-actin-4, HSP90AB1, cytochrome complex, SCP-2 | Mitochondria, MHC II molecules, ICAM-3, phosphatidylserine, sialylated and glycosylated ligands | [28,29,31,35] |
| Nuclear fractions | mRNA and microRNA, other non-coding RNAs | non-coding RNAs | non-coding RNAs | |
| Specific surface markers | Tetraspanins (CD9, CD63, CD 81), ESCRT machinery proteins (Alix, tumor susceptibility gene 10), flotillin-1 | CD40, Phosphatidylserine, integrins, selectins, ESCRT machinery proteins (Alix, Vps4) | Annexin A5, phosphatidylserine, caspase 3, histones | |
| Key functional role | Cell-to-cell communication, cargo | Cell-to-cell communication, cargo | Cell-to-cell communication, cell clearance | |
Basic characteristics of EV subsets
Small EVs
Small EVs are also known as exosomes. They are a derivate of the endocytic membrane with an average diameter of 40-100 nm and are released from several types of cells as a result of exocitosis and production of multi vesicular bodies[18,19]. Multi vesicular bodies move along intracellular tubules, fuse with the plasma membrane and release exosomes into the extracellular space. Small EVs have various cellular components including cytoplasmic and membrane molecules, proteins, hormones (aldosterone), growth factors (vascular endothelial growth factor, transforming growth factor), cytokines [interleukin (IL)-1β, IL-6, IL-8] and lipids, as well as fragments of chromatin, such as non-coding RNAs and several inactive forms of micro RNAs[18,19]. There is also a common set of membrane and cytosolic proteins, which are embedded into exosomes that have originated from distinct cell types[20]. The specific surface markers that ensure recognition of the exosomes are tetraspanins (CD9, CD63, CD81), ESCRT (endosomal sorting complexes required for transport) machinery proteins (Alix, tumor susceptibility gene 10), and flotillin-1[21].
Medium/large EVs
Medium/large EVs (also known as microvesicles, micro particles, ectosomes) range in diameter from 100 to 1000 nm and result from budding of the cell membrane[22]. Medium/large EVs are heavily enriched in phospholipids, such as phosphatidylserine and phosphatidylcholine, and numerous membrane-dependent structures (receptors, CD markers) that originated from the parent cells[23]. Proteomics and lipidomics structure of microvesicles is extremely variable and includes membrane regulatory (Rab, Sterol Carrier Protein 2) and structure (β-actin, α-actin-4) proteins, heat shock proteins HSP90AB1, adhesive molecules (ICAMs, PECAM-1, MCAM), lipids (SpL, PL, LPS, LPS) and receptors (tetraspanin’s receptors, LAIR-1, EGFR), enzymes (superoxide dismutase, Rab GTPase, cytochrome complex, Akt/ERK, triosephosphate isomerase -1, 3-Hydroxy-3-Methylglutaryl-CoA Lyase), immune system proteins (CD14, CD276, MiC-11), and apo-lipoproteins (apo-A-II)[24-26]. Therefore, microvesicles may yield several non-coding RNAs and chromatin fragments coupled with the complexity of other components[27].
Apoptotic cell-derived EVs
Apoptotic cell-derived EVs include two types of apoptotic bodies: large membrane-bound vesicles [large apoptotic bodies (ABs) with diameter ≥ 1000 nm] and small apoptotic microvesicles (small ABs with diameter < 1000 nm)[28]. ABs are particles that are generally larger in size in comparison to both exosomes and microvesicles but have a variable diameter that fluctuates around 1000 nm (from 1000 nm to 2000 nm)[29]. Both types of ABs result from blebbing of the surface of apoptotic cells and contain proteins, numerous cell organelles and chromatin fractions, such as non-coding RNAs from the nucleus or nucleoli[30]. The process of AB generation is controlled by several distinct morphological steps (i.e., membrane permeability and blebs, membrane protrusion, and cell fragmentation), which are, in turn, regulated by several molecular factors including the Rho-associated protein kinase and the plasma membrane channel pannexin-1.
ABs contain mitochondria, MHC II molecules, ICAM-3, phosphatidylserine, sialylated and glycosylated ligands, fragments of chromatin, DNAs, and non-coding RNAs. It has been noted that the packaging of chromatin content (DNAs and non-coding RNAs) into the structure of ABs is regulated by apoptosis and there are indeed, ABs with no fragments of chromatin or very low amounts of DNAs[31]. ABs are also classified depending on their origin from the mother cells including antigen-presenting cells, mononuclear cells, endothelial cells, fibroblasts, cardiac myocytes, and epithelial cells[32]. The clearance of ABs has been ensured by phagocytes[33]. To accurately differentiate ABs from other particles including cells and debris, there are several specific surface markers such as Annexin A5/phosphatidylserine[34].
Biological role of EVs
The key biological functions of EVs that originate from various cells are cell-to-cell communication and the transfer of materials called the secretome. Acting as cargo for numerous molecules [heat shock proteins (HSP-90, HSP-70), ILs, tumor necrosis factor-alpha, active molecules, enzymes, peptides, growth factors], EVs are recognized by target cells through specific antigens, bind and fuse with them to supply the packaged materials within to the cells. Therefore, small and medium/large EVs have wide range of biological functions including immune response, antigen presentation, and the transfer of RNA and DNA[29,35]. The full spectrum of pleiotropic effects of circulating EVs is shown in Figure 1.
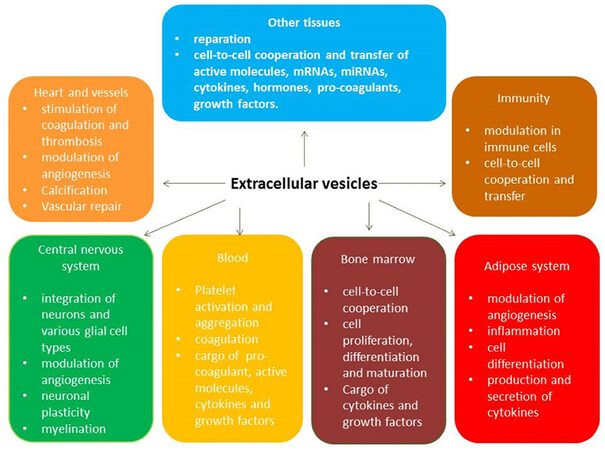
Figure 1. Pleiotropic effects of extracellular vesicles
Recent studies have revealed that EVs may contain inactive forms of non-coding RNAs, which can be transferred to another cell and become functional in that new microenvironment[36,37]. Indeed, there is strong evidence that hypoxia and ischemia are triggers for monocyte-dependent production of pro-inflammatory cytokines including IL-2 and TNF-alpha, and the supply of these cytokines to target cells are mediated through package as cargo into EVs[38]. On the other hand, HSPs, growth factors, non-coding RNAs, and active molecules, which are all transferred by EVs, are involved in the regulation of reparative response, immune reactions and cytoprotection[39,40]. The wide spectrum of biologically active molecules that are transported by EVs from the mother cells to target cells are able to regulate the endogenous repair system activity including proliferation, differentiation and migration of endothelial progenitor cells and angiogenesis[41,42]. Through appropriate receptor-ligand (integrin αvβ3, CD40 ligand, neuregulin-1, VE-cadherin and beta-catenin) interactions and the cargo content of EVs, the intracellular signaling pathways can be regulated to ensure activation of endothelial cells as well as the attraction and internalization of various types of circulating blood cells (platelets, mononuclear cells, macrophages, lymphocytes) by the endothelial cell surface[42]. Moreover, vascular growth, restoration of vascular integrity and function, as well as the recruitment of inflammatory cells, may all be directly related to up-regulated expression of neuregulin-1 in endothelial cells as a result of EV-dependent stimulation, because circulating EVs can be a source of a variety of pro-angiogenic mRNAs including neuregulin-1 mRNA[43]. Additionally, EVs may induce a cytoskeleton-junction response from endothelial cells that is characterized by myosin light chain phosphorylation, contractile fiber reorganization, VE-cadherin phosphorylation and adherent junction dissociation. This process is a key mechanism of increasing permeability of the vascular wall, releasing neutrophil extracellular traps containing citrullinated histones and myeloperoxidase, and in developing senescence and acceleration of atherosclerosis[44-46]. The proteome of EVs consists of pro-coagulant components such as tissue factor and phospholipids, which play a pivotal role in coagulation and the triggering of vasoocclusion in CAD[47,48].
EVs in various stages of atherosclerosis development
Modification of macrophages’ phenotype and function
Macrophages are the primary antigen-presenting cells in atherosclerotic lesions and provide the fundamental link between microvascular inflammation and atherosclerotic plaque development and progression. It has been suggested that endothelial cell-derived EVs export microRNA-92a from mother cells to macrophages in response to atheroprone stimuli to change macrophage phenotype, regulate their functions and enhance atherosclerotic plaque shaping[49]. Indeed, over-expression of microRNA-92a in endothelial cells in atherosclerosis enhances the pro-inflammatory response in the vasculature, supports low-density lipoprotein (LDL) uptake, and impairs the migration of macrophages through changes in their phenotype from atheroprotected to atheroprone[50]. Interestingly, the expression of microRNA-92a in mature endothelial cells is up-regulated by the combination of several factors such as low shear stress, atherogenic oxidized LDL, IL-6, atheroprotective Kruppel-like factor (KLF)-2 and KLF-4, and suppressor of cytokine signaling 5[51,52]. The expression of pro-inflammatory cytokine-induced markers such as monocyte chemotactic protein 1, vascular cell adhesion molecule-1 (VCAM-1), E-selectin, VE-cadherin, and endothelial nitric oxide synthase, as well as KLF-2 and KLF-4, all strongly correspond to down-regulated microRNA-92a in the endothelium in animals[53]. Therefore, atherosclerosis induces oxidized LDL, and KLF-2 regulates the expression of inflammation-associated microRNA-155 in endothelial cells[53]. Moreover, it has been found that endothelial cell-derived EVs enriched in oxidized LDL and microRNA-155 influenced monocyte activation by shifting the monocytes/macrophages balance in the vasculature from the anti-inflammatory M2 phenotype of macrophages to the pro-inflammatory M1 phenotype of macrophages[52,53]. Accumulation of macrophages with the M1 phenotype in the vascular wall also ensured a link between microvascular inflammation and impaired vasodilatory responses to flow via the regulation of microRNA-92-dependent presentation of KLF-2 and oxidative stress stimulation[54,55]. Additionally, endothelial cell-derived EVs that were packaged with pyruvate kinase muscle isozyme 2 triggered re-programming of B cells and the activation of T cells via its cargo of interferon-gamma[56]. This mechanism was found to be an important element for the suppression of mononuclear transformation into macrophages with the inflammatory phenotype. The total number of endothelial cell-derived EVs was significantly and positively correlated with oxidative stress and systemic inflammation in healthy younger individuals. While the ability of activated endothelial cells to release EVs packed with pro-angiogenic molecules progressively decreases in patients with established CAD, apoptotic endothelial cell-derived EVs appear to be detected in higher concentrations[57,58]. This phenomenon probably reflects maladaptive responses of the endothelium in advanced atherosclerosis and decreased control of local vascular inflammation is associated with an altered, intra-plaque immune phenotype of the cells including macrophages and endothelial cells [Figure 2]. It is not clear whether local vascular injury appears first or the alteration in gene regulation of pro-inflammatory genes emerges initially as a microvascular response, thereby triggering acceleration of atherosclerosis.
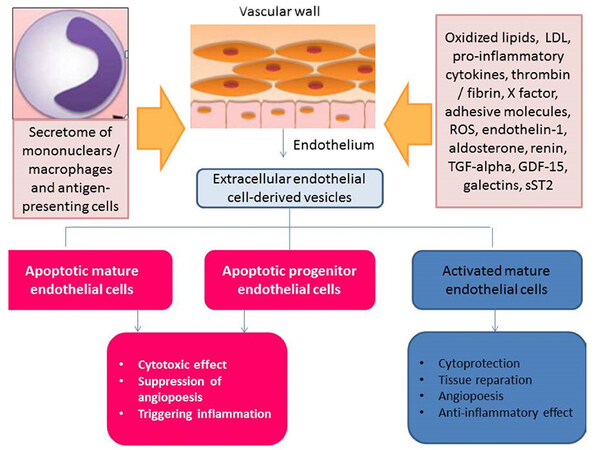
Figure 2. The controversial roles of apoptotic endothelial cell-derived EVs and activated endothelial cell-derived EVs in vascular homeostasis. EVs: extracellular vesicles; LDL: low-density lipoproteins; ROS: reactive oxide species; TGF: transforming growth factor; GDF-15: growth-differential factor-15; sST2: soluble suppressor tumorigenisity-2
Mediating angiogenesis and neovascularization
Endothelial cell-derived EVs are involved in the regulation of vascular function, integrity and angiogenesis through transportation of a wide spectrum of micro-RNA (microRNA-126-3p, microRNA-222-3p, microRNA-let-7d-5p, microRNA-21-5p, microRNA-26a-5p, microRNA-92a-3p, microRNA-139-5p, microRNA-30b-5p, microRNA-150 and microRNA-199a-5p) that have been implicated in MAPK/ERK1/2, c-Jun N-terminal kinases/stress-activated kinases signaling cascade, and nuclear factor-κB signaling pathway to target cells [progenitor and mature endothelial cells, macrophages, smooth muscle cells (SMC), fibroblasts] in arterial endothelium[51,52,59,60]. There is evidence that the expression of thrombospondin 1 (THBS1), an inhibitor of angiogenesis and a target for microRNA-92a-3p, was significantly up-regulated in the endothelial cells in atherosclerosis and that endothelial cell-derived EV transferred microRNA-92a-3p to reduce THBS1 expression[52]. Interestingly, the number of activated endothelial cell-derived EVs decreased, whereas the number of apoptotic endothelial cell-derived EVs increased in patients with atherosclerosis when compared with healthy individuals. This discrepancy was found to be closely associated with impaired repair in atherosclerotic injury of the vasculature[61].
Plaque formation and vascular calcification
Endothelial cell-derived EVs contain a wide spectrum of intercellular signaling molecules, enzymes, regulatory proteins, and growth factors, all of which are involved in the regulation of plaque formation and vascular calcification[61,62]. Transforming growth factor-beta (TGF-β), which induces the expression of von Willebrand Factor, proliferation of SMCs and progenitor endothelial cells, and recruitment of monocytes/macrophages, is transferred by activated endothelial cell-derived EVs[63,64]. There is a wide range of evidence that pro-inflammatory cytokines including tumor necrosis factor-alpha and some adipocytokines (adiponectin) induce VCAM-1 production in mature endothelial cells, which is then accompanied by enhanced circulating leukocyte attachment and a weakened ability to release EVs after activation[65,66]. Therefore, the cellularity of the plaque can be modified by signaling molecules transferred by EVs. Indeed, deficiencies in endothelial microRNA-126 and microRNA-92a transmitted as cargo with endothelial cell-derived EVs has demonstrated acceleration of neointimal lesion formation of carotid arteries, increased smooth muscle cell turnover, and its release was reduced by atheroprotective laminar shear stress[67-69]. It has been established that autophagy of endothelial cells, fibroblasts, and CD45+ hematopoietic cells that accumulates into an atheroma plays a certain role in shaping vulnerable plaques and act as a trigger for plaque rupture[70]. There were intriguing findings that clarify a role of down-regulated microRNA-92a-3p in endothelial cell autophagy through de-repressed autophagy-related gene 4a and increasing activity of luciferase in autophagy-related gene 4a containing 3’UTR[71,72]. Moreover, EV-derived microRNA-92a-3p upregulates the expression of cell cycle and mitosis-related genes including claudin-11, and downregulates the adhesion-related gene expression in endothelial and foam cells[73]. Thus, the packaging and transfer of microRNA-92a-3p by endothelial cell-derived EVs ensures both a protective effect and induces repair of the endothelium, thereby preventing plaque rupture and intravascular thrombosis[74].
The mediation of osteogenic trans-differentiation of vascular SMCs by inflammation, endothelial dysfunction and reactive oxygen species by EVs may be considered as the key regulators of vascular and plaque calcification. EVs also participate in the formation of microvascular calcifications that are implicated in atherosclerotic plaque formation and rupture[75,76]. Interestingly, the release of exosomes by EVs may promote microvascular calcification in response to environmental calcium stress[75]. Sortilin, which is a key regulator of SMC calcification through its recruitment to EVs[76], has been found to regulate loading of the calcification protein - tissue nonspecific alkaline phosphatase - into EVs and thereby confer calcification potency. SMC calcification also requires Rab11-dependent trafficking and FAM20C/casein kinase 2-dependent C-terminal phosphorylation of sortilin; the deficiency of sortilin was thus found to suppress ectopic vascular calcification[75,76]. Although it is not clear how EVs influence vascular calcification, previous clinical studies have shown that cell plaque composition, volume of the plaque, a lipid core and the Agatston coronary calcium score were all inversely correlated with the number of endothelial cell-derived EVs[77]. However, a recent clinical study based on positron emission tomography/computed tomography imaging has shown that early microvascular calcifications can be identified frequently, even in high-risk patients with plaques[78]. Early microvascular calcification was found to be associated with atherosclerotic plaque instability and rupture, whereas advanced macrovascular calcification can potentially contribute to plaque stability[79,80]. There is a suggestion that M1 macrophages that have accumulated in a plaque may release EVs enriched in S100A9 and annexin A5 as a result of weakly activated endothelial cell-derived EVs’ stimulation. This contributes to accelerated trans-differentiation of SMCs into osteogenic cells, and the potentiation of microvascular calcification[79]. Notably, endothelial cell-derived EVs contain bone-related matricellular proteins (osteopontin, osteonectin, osteoprotegerin) and the deficiency of these EVs in the circulation may impair the migration of and cell fusion required for osteoclast formation in the vasculature[81,82]. Moreover, the immune phenotype and the number of cells accumulating into an atheroma, as well as the extracellular environment are all under the control of endothelial cell-derived EVs[83]. In fact, EVs originating from progenitor and mature endothelial cells have the unique ability of regulating the osteogenic transformation of SMCs and the activation of fibroblasts in the vasculature. The endothelial cell-derived EVs support microvascular calcification in the collagen-poor fibrous cap, and promote plaque rupture by acting through the TGF-β/SMAD signaling and platelet-derived growth factor-BB pathway[84]. Interestingly, endothelial cell senescence may increase the release of EVs as carriers of molecular information, which then contributes to the development and calcification of atherosclerotic plaques. The role of senescent EVs in microvascular calcification is not certain however, and requires further investigation[85]. Whether the altered balance between EVs produced by activated and apoptotic EVs is a cause of microvascular calcification, or if the adaptive reaction prevents inflammatory-related injury of the vasculature and thereby reduce the risk of calcification, is not fully understood.
Endothelial dysfunction and EVs
Endothelial cell-derived EVs are established biomarkers of endothelial dysfunction[86]. The interplays between cell components that are embedded into the pathogenesis of vascular tone impairment and vascular remodeling in atherosclerosis are mutually activated and sophisticated. There is a wide range of evidence that foam cells and both progenitor and mature endothelial cells may work with each other by releasing exosomes and ABs. Foam cells may secrete exosomes that suppress endogenous activity of endothelial cells and their precursors to modulate endothelial-dependent vasodilatation and prevent intravascular blood cell adhesion and thrombosis[87]. Moreover, the foam cell-derived EVs promote migration and proliferation of SMCs by regulating the actin cytoskeleton and focal adhesion via ERK and Akt pathways, thereby acting as a trigger of atherosclerosis[87].
There are findings that demonstrate a causative impact of EV-packaged microRNA-145, microRNA-150 and microRNA-126 on the progression of endothelial dysfunction and atherosclerosis in vivo[88]. These microRNAs appear to respect tissue specificity and are both expressed in and released from endothelial cells due to several stimuli including shear stress, inflammatory cytokines, cell adhesion and thrombosis. Down-regulated microRNA-145, which plays a key role in the control of SMC differentiation, promotes lesion formation. The endothelial cell-specific microRNA-126 is a powerful signal transducer, which is essential for endothelial repair through its transfer from apoptotic endothelial cells derived EVs. Interestingly, splicing of the X-box binding protein 1 (XBP1) in vascular SMCs may control endothelial cell migration via EVs-mediated transfer of microRNA-150 and microRNA-150-driven vascular endothelial growth factor-dependent PI3K/Akt pathway activation, thereby supporting homeostasis of the vasculature[89]. In fact, XBP1 deficiency in vascular SMCs and endothelial progenitor cells significantly attenuate angiogenesis and neovascularization, as well as maintain endothelial integrity and resistance to apoptosis[90,91]. Foam cell shaping driven by CD36 mediated internalization of oxidized LDL activates mononuclear cells and endothelial cells, and the subsequent release of EVs embedded with pro-inflammatory leukotriene B4, which promotes endothelial dysfunction and accelerates atherosclerosis through the high-affinity receptor BLTR1[92]. In fact, the presence of dysregulated inflammatory molecules on the surface of the endothelial cell layer was associated with increased coagulation due to over-expression of the glycoprotein (GP) IIb/IIIa (integrin αIIbβ3) receptor, anomalous clot formation or shaping amyloid fibrin[93,94]. Importantly, the GPIIb/IIIa receptors were additionally shed into EVs and transferred as cargo to the target cells for translation of activation signals remotely. Another finding has demonstrated that heat shock protein 27 (HSP27) packaged into EVs via activating TLR-4/NF-κB in the target cells can attenuate endothelial function, reduce vascular and plaque inflammation, lower cholesterol levels and suppress atherogenesis in animal models[95]. However, the angiopoetic role of activated endothelial cell-derived EVs is not always considered as having a positive impact on the endothelium. For instance, neovascularization of the shoulder region of the plaque’s cap was associated with instability of the atheroma due to the increased risk of rupture[94,96]. There are several excellent reviews that are dedicated the role of EVs in vascular homeostasis and its relation to CV disease development[97,98]. Thus, EVs promote the function of target cells through the transfer of surface integrins and receptors, cellular fusion and the delivery of various active molecules.
EVs as diagnostic and predictive biomarkers of atherosclerosis
The diagnostic and predictive roles of endothelial cell-derived EVs in atherosclerosis and MACEs are uncertain[99]. However, there has been progress in the diagnosis, prognostication and treatment of CV diseases with EVs[100]. For instance, HSP27 packaged in endothelial cell-derived EVs was found to be a predictor of a lower CV risk among patients having a heart attack, stroke, or death from CV disease[95]. The imbalance between activated and apoptotic endothelial cell-derived EVs has provided additional prognostic information for patients with established CV disease, including acute myocardial infarction, acute coronary syndrome, ischemic heart failure, MACEs, and arrhythmias, as well as individuals with metabolic diseases having a higher risk of CV events and disease[101-104].
Future directions toward the role of EVs in atherosclerosis
EVs have demonstrated a pivotal role in transferring numerous bioactive molecules, supporting cell-to-cell cooperation, and regulate gene expression in target cells. EVs-based therapeutic regenerative strategies may thus be used to attenuate tissue injury and promote vascular regeneration and repair[105,106]. Accumulating evidence implicates EVs in the development and progression of atherosclerosis, and creates the possibility of using EVs for personalized therapeutic strategies. Therefore, single-EV analysis may identify signatures of exosome-derived DNA/non-coding RNAs including microRNA, regulator proteins, and other components both as diagnostic and predictive biomarkers in atherosclerosis[107]. Large clinical studies are required for further elucidation of whether EVs can be excellent options for point-of-care diagnosis and individual treatment.
Conclusion
EVs could be promising biomarkers with both diagnostic and predictive values, while their number, content, immune phenotype and origin may provide more useful information about the pathophysiology of atherosclerosis and help stratify patients at risk of MACEs. The emergence of endothelial cell-derived EVs provide favorable and promising strategies, not only for CV risk stratification in vulnerable populations but for individualized treatment of atherosclerosis and other CV diseases.
Declarations
Authors’ contributionsParticipated in drafting the article, revised it critically for important intellectual content: Berezin AE
Gave final approval of the version to be submitted: Berezin AA
Both authors make eqaual contributions to conception and design, searchig data, analysis, interpretation of data and writting of the manuscript.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res 2016;118:535-46.
2. Barquera S, Pedroza-Tobías A, Medina C, Hernández-Barrera L, Bibbins-Domingo K, et al. Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch Med Res 2015;46:328-38.
3. Moran AE, Tzong KY, Forouzanfar MH, Rothy GA, Mensah GA, et al. Variations in ischemic heart disease burden by age, country, and income: the Global Burden of Diseases, Injuries, and Risk Factors 2010 study. Glob Heart 2014;9:91-9.
4. Cairns BJ, Balkwill A, Canoy D, Green J, Reeves GK, et al; Million Women Study Collaborators. Variations in vascular mortality trends, 2001-2010, among 1.3 million women with different lifestyle risk factors for the disease. Eur J Prev Cardiol 2015;22:1626-34.
5. Li K, Hüsing A, Kaaks R. Lifestyle risk factors and residual life expectancy at age 40: a German cohort study. BMC Med 2014;12:59.
6. Gimbrone MA Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res 2016;118:620-36.
7. Horio E, Kadomatsu T, Miyata K, Arai Y, Hosokawa K, et al. Role of endothelial cell-derived angptl2 in vascular inflammation leading to endothelial dysfunction and atherosclerosis progression. Arterioscler Thromb Vasc Biol 2014;34:790-800.
8. Stoll G, Bendszus M. Inflammation and atherosclerosis: novel insights into plaque formation and destabilization. Stroke 2006;37:1923-32.
9. Libby P, Bornfeldt KE, Tall AR. Atherosclerosis: successes, surprises, and future challenges. Circ Res 2016;118:531-4.
10. Rader DJ. Human genetics of atherothrombotic disease and its risk factors. Arterioscler Thromb Vasc Biol 2015;35:741-7.
11. Dron JS, Ho R, Hegele RA. Recent advances in the genetics of atherothrombotic disease and its determinants. Arterioscler Thromb Vasc Biol 2017;37:e158-66.
12. Boulanger CM, Loyer X, Rautou PE, Amabile N. Extracellular vesicles in coronary artery disease. Nat Rev Cardiol 2017;14:259-72.
13. Nguyen MA, Karunakaran D, Geoffrion M, Cheng HS, Tandoc K, et al. Extracellular vesicles secreted by atherogenic macrophages transfer microRNA to inhibit cell migration. Arterioscler Thromb Vasc Biol 2018;38:49-63.
14. Deng W, Tang T, Hou Y, Zeng Q, Wang Y, et al. Extracellular vesicles in atherosclerosis. Clin Chim Acta 2019;495:109-17.
15. Hafiane A, Daskalopoulou SS. Extracellular vesicles characteristics and emerging roles in atherosclerotic cardiovascular disease. Metabolism 2018;85:213-22.
16. Boilard E. Extracellular vesicles and their content in bioactive lipid mediators: more than a sack of microRNA. J Lipid Res 2018;59:2037-46.
17. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 2018;7:1535750.
18. Clayton A, Boilard E, Buzas EI, Cheng L, Falcón-Perez JM, et al. Considerations towards a roadmap for collection, handling and storage of blood extracellular vesicles. J Extracell Vesicles 2019;8:1647027.
19. Hill AF, Pegtel DM, Lambertz U, Leonardi T, O’Driscoll L, et al. ISEV position paper: extracellular vesicle RNA analysis and bioinformatics. J Extracell Vesicles 2014;2:22859.
20. Raimondo F, Morosi L, Chinello C, Magni F, Pitto M. Advances in membranous vesicle and exosome proteomics improving biological understanding and biomarker discovery. Proteomics 2011;11:709-20.
21. Corrado C, Raimondo S, Chiesi A, Ciccia F, De Leo G, et al. Exosomes as intercellular signaling organelles involved in health and disease: basic science and clinical applications. Int J Mol Sci 2013;14:5338-66.
22. Tual-Chalot S, Leonetti D, Andriantsitohaina R, Martinez MC. Microvesicles: intercellular vectors of biological messages. Mol Interv 2011;11:88-94.
23. Soo CY, Song YQ, Zheng Y, Campbell EC, Riches AC, et al. Nanoparticle tracking analysis monitors microvesicle and exosome secretion from immune cells. Immunology 2012;136:192-7.
24. Huang XY, Yuan TZ, Tschannen M, Sun ZF, Jacob H, et al. Characterization of human plasma-derived exosomal rnas by deep sequencing. BMC Genomics 2013;14:14.
25. de Jong OG, Verhaar MC, Chen Y, Vader P, Gremmels H, et al. Cellular stress conditions are reflected in the protein and rna content of endothelial cell-derived exosomes. J Extracell Vesicles 2012;1.
26. Waldenstrom A, Genneback N, Hellman U, Ronquist G. Cardiomyocyte microvesicles contain dna/rna and convey biological messages to target cells. PLoS One 2012;7:7.
27. Camussi G, Deregibus MC, Bruno S, Grange C, Fonsato V, et al. Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am J Cancer Res 2011;1:98-110.
28. Caruso S, Poon IKH. Apoptotic cell-derived extracellular vesicles: more than just debris. Front Immunol 2018;9:1486.
29. Boon RA, Vickers KC. Intercellular transport of micrornas. Arterioscler Thromb Vasc Biol 2013;33:186-92.
30. Gump JM, Thorburn A. Autophagy and apoptosis: what is the connection? Trends Cell Biol 2011;21:387-92.
31. Jiang L, Paone S, Caruso S, Atkin-Smith GK, Phan TK, et al. Determining the contents and cell origins of apoptotic bodies by flow cytometry. Sci Rep 2017;7:14444.
32. Akers JC, Gonda D, Kim R, Carter BS, Chen CC. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol 2013;113:1-11.
33. Orlando KA, Stone NL, Pittman RN. Rho kinase regulates fragmentation and phagocytosis of apoptotic cells. Exp Cell Res 2006;312:5-15.
34. Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, et al. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol 2008;445:29-76.
35. Simpson RJ, Lim JW, Moritz RL, Mathivanan S. Exosomes: proteomic insights and diagnostic potential. Expert Rev Proteomics 2009;6:267-83.
36. Mause SF, Weber C. Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ Res 2010;107:1047-57.
37. El Andaloussi S, Maeger I, Breakefield XO, Wood MJA. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat Rev Drug Discov 2013;12:348-58.
38. Yu X, Deng LY, Wang D, Li N, Chen X, et al. Mechanism of TNF-alpha autocrine effects in hypoxic cardiomyocytes: Initiated by hypoxia inducible factor 1 alpha, presented by exosomes. J Mol Cell Cardiol 2012;53:848-57.
39. Tian J, Guo X, Liu XM, Liu L, Weng QF, et al. Extracellular hsp60 induces inflammation through activating and up-regulating TLRs in cardiomyocytes. Cardiovasc Res 2013;98:391-401.
40. Hoyer FF, Nickenig G, Werner N. Microparticles - messengers of biological information. J Cell Mol Med 2010;14:2250-6.
41. Todorova D, Simoncini S, Lacroix R, Sabatier F, Dignat-George F. Extracellular vesicles in angiogenesis. Circ Res 2017;120:1658-73.
42. Bagi Z, Couch Y, Broskova Z, Perez-Balderas F, Yeo T, et al. Extracellular vesicle integrins act as a nexus for platelet adhesion in cerebral microvessels. Sci Rep 2019;9:15847.
43. Figliolini F, Ranghino A, Grange C, Cedrino M, Tapparo M, et al. Extracellular vesicles from adipose stem cells prevent muscle damage and inflammation in a mouse model of hind limb ischemia: role of neuregulin-1. Arterioscler Thromb Vasc Biol 2020;40:239-54.
44. Chatterjee V, Yang X, Ma Y, Cha B, Meegan JE, et al. Endothelial microvesicles carrying Src-rich cargo impair adherens junction integrity and cytoskeleton homeostasis. Cardiovasc Res 2019:cvz238.
45. Berezin A. Neutrophil extracellular traps: The core player in vascular complications of diabetes mellitus. Diabetes Metab Syndr 2019;13:3017-23.
46. Dalli J, Montero-Melendez T, Norling LV, Yin X, Hinds C, et al. Heterogeneity in neutrophil microparticles reveals distinct proteome and functional properties. Mol Cell Proteomics 2013;12:2205-19.
47. Banfi C, Brioschi M, Wait R, Begum S, Gianazza E, et al. Proteome of endothelial cell-derived procoagulant microparticles. Proteomics 2005;5:4443-55.
48. Camus SM, De Moraes JA, Bonnin P, Abbyad P, Le Jeune S, et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vaso occlusions in sickle cell disease. Blood 2015;125:3805-14.
49. Chang YJ, Li YS, Wu CC, Wang KC, Huang TC, et al. Extracellular microRNA-92a mediates endothelial cell-macrophage communication. Arterioscler Thromb Vasc Biol 2019;39:2492-504.
50. Loyer X, Potteaux S, Vion AC, Guérin CL, Boulkroun S, et al. Inhibition of microRNA-92a prevents endothelial dysfunction and atherosclerosis in mice. Circ Res 2014;114:434-43.
51. Liu Y, Li Q, Hosen MR, Zietzer A, Flender A, et al. Atherosclerotic conditions promote the packaging of functional microRNA-92a-3p into endothelial microvesicles. Circ Res 2019;124:575-87.
52. Fang Y, Davies PF. Site-specific microRNA-92a regulation of Kruppel-like factors 4 and 2 in atherosusceptible endothelium. Arterioscler Thromb Vasc Biol 2012;32:979-87.
53. He S, Wu C, Xiao J, Li D, Sun Z, et al. Endothelial extracellular vesicles modulate the macrophage phenotype: Potential implications in atherosclerosis. Scand J Immunol 2018;87:e12648.
54. Wu W, Xiao H, Laguna-Fernandez A, Villarreal G Jr, Wang KC, et al. Flow-dependent regulation of kruppel-like factor 2 is mediated by microRNA-92a. Circulation 2011;124:633-41.
55. Gou L, Zhao L, Song W, Wang L, Liu J, et al. Inhibition of miR-92a Suppresses oxidative stress and improves endothelial function by upregulating heme oxygenase-1 in db/db Mice. Antioxid Redox Signal 2018;28:358-70.
56. Yang J, Dang G, Lü S, Liu H, Ma X, et al. T-cell-derived extracellular vesicles regulate B-cell IgG production via pyruvate kinase muscle isozyme 2. FASEB J 2019;33:12780-99.
57. Žėkas V, Matuzevičienė R, Karčiauskaitė D, Mažeikienė A, Burokienė N, et al. Chronic and oxidative stress association with total count of endothelial microvesicles in healthy young male plasma. Adv Clin Exp Med 2019;28:683-92.
58. Berezin AE, Kremzer AA, Samura TA, Berezina TA. Altered signature of apoptotic endothelial cell-derived microvesicles predicts chronic heart failure phenotypes. Biomark Med 2019;13:737-50.
59. Wang Z, Zhang J, Zhang S, Yan S, Wang Z, et al. MiR-30e and miR-92a are related to atherosclerosis by targeting ABCA1. Mol Med Rep 2019;19:3298-304.
60. Iaconetti C, Polimeni A, Sorrentino S, Sabatino J, Pironti G, et al. Inhibition of miR-92a increases endothelial proliferation and migration in vitro as well as reduces neointimal proliferation in vivo after vascular injury. Basic Res Cardiol 2012;107:296.
61. Kang S, Yang JW, Jeong JY, Park J, An HJ, et al. Size distribution of serum extracellular vesicles in mice with atherosclerosis. Pathol Res Pract 2019;215:152574.
62. Yang W, Zou B, Hou Y, Yan W, Chen T, et al. Extracellular vesicles in vascular calcification. Clin Chim Acta 2019;499:118-22.
63. Chimen M, Evryviadou A, Box CL, Harrison MJ, Hazeldine J, et al. Appropriation of GPIbα from platelet-derived extracellular vesicles supports monocyte recruitment in systemic inflammation. Haematologica 2019:haematol.2018.215145.
64. Wang Y, Xie Y, Zhang A, Wang M, Fang Z, et al. Exosomes: an emerging factor in atherosclerosis. Biomed Pharmacother 2019;115:108951.
65. Wadey RM, Connolly KD, Mathew D, Walters G, Rees DA, et al. Inflammatory adipocyte-derived extracellular vesicles promote leukocyte attachment to vascular endothelial cells. Atherosclerosis 2019;283:19-27.
66. Blaser MC, Aikawa E. Differential miRNA loading underpins dual harmful and protective roles for extracellular vesicles in atherogenesis. Circ Res 2019;124:467-9.
67. Wang J, Zhang C, Li C, Zhao D, Li S, et al. MicroRNA-92a promotes vascular smooth muscle cell proliferation and migration through the ROCK/MLCK signalling pathway. J Cell Mol Med 2019;23:3696-710.
68. Zhou J, Li YS, Nguyen P, Wang KC, Weiss A, et al. Regulation of vascular smooth muscle cell turnover by endothelial cell-secreted microRNA-126: role of shear stress. Circ Res 2013;113:40-51.
69. Liu H, Li G, Zhao W, Hu Y. Inhibition of miR-92a may protect endothelial cells after acute myocardial infarction in rats: role of KLF2/4. Med Sci Monit 2016;22:2451-62.
70. Zhang YY, Shi YN, Zhu N, Wang W, Deng CF, et al. Autophagy: a killer or guardian of vascular smooth muscle cells. J Drug Target 2020:1-7.
71. Rogg EM, Abplanalp WT, Bischof C, John D, Schulz MH, et al. Analysis of cell type-specific effects of microRNA-92a provides novel insights into target regulation and mechanism of action. Circulation 2018;138:2545-58.
72. Daniel JM, Penzkofer D, Teske R, Dutzmann J, Koch A, et al. Inhibition of miR-92a improves re-endothelialization and prevents neointima formation following vascular injury. Cardiovasc Res 2014;103:564-72.
73. Yamada NO, Heishima K, Akao Y, Senda T. Extracellular vesicles containing microRNA-92a-3p facilitate partial endothelial-mesenchymal transition and angiogenesis in endothelial cells. Int J Mol Sci 2019;20:E4406.
74. Barbati C, Vomero M, Colasanti T, Ceccarelli F, Marcosano M. Microparticles and autophagy: a new frontier in the understanding of atherosclerosis in rheumatoid arthritis. Immunol Res 2018;66:655-62.
75. Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res 2015;116:1312-23.
76. Goettsch C, Hutcheson JD, Aikawa M, Iwata H, Pham T, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest 2016;126:1323-36.
77. Chiva-Blanch G, Padró T, Alonso R, Crespo J, Perez de Isla L, et al. Liquid biopsy of extracellular microvesicles maps coronary calcification and atherosclerotic plaque in asymptomatic patients with familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2019;39:945-55.
78. Creager MD, Hohl T, Hutcheson JD, Moss AJ, Schlotter F, et al. 18F-fluoride signal amplification identifies microcalcifications associated with atherosclerotic plaque instability in positron emission tomography/computed tomography images. Circ Cardiovasc Imaging 2019;12:e007835.
79. Adamson PD, Vesey AT, Joshi NV, Newby DE, Dweck MR. Salt in the wound: (18)F-fluoride positron emission tomography for identification of vulnerable coronary plaques. Cardiovasc Diagn Ther 2015;5:150-5.
80. Dweck MR, Chow MW, Joshi NV, Williams MC, Jones C, et al. Coronary arterial 18F-sodium fluoride uptake: a novel marker of plaque biology. J Am Coll Cardiol 2012;59:1539-48.
81. New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, et al. Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res 2013;113:72-7.
82. Sodek J, Zhu B, Huynh MH, Brown TJ, Ringuette M. Novel functions of the matricellular proteins osteopontin and osteonectin/SPARC. Connect Tissue Res 2002;43:308-19.
83. Martins M, Ribeiro D, Martins A, Reis RL, Neves NM. Extracellular vesicles derived from osteogenically induced human bone marrow mesenchymal stem cells can modulate lineage commitment. Stem Cell Reports 2016;6:284-91.
84. Krohn JB, Hutcheson JD, Martínez-Martínez E, Irvin WS, Bouten CV, et al. Discoidin domain receptor-1 regulates calcific extracellular vesicle release in vascular smooth muscle cell fibrocalcific response via transforming growth factor-β signaling. Arterioscler Thromb Vasc Biol 2016;36:525-33.
85. Alique M, Ramírez-Carracedo R, Bodega G, Carracedo J, Ramírez R. Senescent Microvesicles: a novel advance in molecular mechanisms of atherosclerotic calcification. Int J Mol Sci 2018;19:E2003.
86. Blaser MC, Aikawa E. Roles and regulation of extracellular vesicles in cardiovascular mineral metabolism. Front Cardiovasc Med 2018;5:187.
87. Paone S, Baxter AA, Hulett MD, Poon IKH. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell Mol Life Sci 2019;76:1093-106.
88. Wei Y, Nazari-Jahantigh M, Neth P, Weber C, Schober A. MicroRNA-126, -145, and -155: a therapeutic triad in atherosclerosis? Arterioscler Thromb Vasc Biol 2013;33:449-54.
89. Zhao Y, Li Y, Luo P, Gao Y, Yang J, et al. XBP1 splicing triggers miR-150 transfer from smooth muscle cells to endothelial cells via extracellular vesicles. Sci Rep 2016;6:28627.
90. Yin M, Loyer X, Boulanger CM. Extracellular vesicles as new pharmacological targets to treat atherosclerosis. Eur J Pharmacol 2015;763:90-103.
91. Togliatto G, Dentelli P, Rosso A, Lombardo G, Gili M, et al. PDGF-BB carried by endothelial cell-derived extracellular vesicles reduces vascular smooth muscle cell apoptosis in diabetes. Diabetes 2018;67:704-16.
92. Sanden M, Botha J, Nielsen MRS, Nielsen MH, Schmidt EB, et al. BLTR1 and CD36 expressing microvesicles in atherosclerotic patients and healthy individuals. Front Cardiovasc Med 2018;5:156.
93. Pretorius L, Thomson GJA, Adams RCM, Nell TA, Laubscher WA, et al. Platelet activity and hypercoagulation in type 2 diabetes. Cardiovasc Diabetol 2018;17:141.
94. Vítková V, Živný J, Janota J. Endothelial cell-derived microvesicles: potential mediators and biomarkers of pathologic processes. Biomark Med 2018;12:161-75.
95. Shi C, Ulke-Lemée A, Deng J, Batulan Z, O’Brien ER. Characterization of heat shock protein 27 in extracellular vesicles: a potential anti-inflammatory therapy. FASEB J 2019;33:1617-30.
96. Jansen F, Li Q, Pfeifer A, Werner N. Endothelial- and immune cell-derived extracellular vesicles in the regulation of cardiovascular health and disease. JACC Basic Transl Sci 2017;2:790-807.
97. Hutcheson JD, Aikawa E. Extracellular vesicles in cardiovascular homeostasis and disease. Curr Opin Cardiol 2018;33:290-7.
98. Jansen F, Nickenig G, Werner N. Extracellular vesicles in cardiovascular disease: potential applications in diagnosis, prognosis, and epidemiology. Circ Res 2017;120:1649-57.
99. Pardo F, Villalobos-Labra R, Sobrevia B, Toledo F, Sobrevia L. Extracellular vesicles in obesity and diabetes mellitus. Mol Aspects Med 2018;60:81-91.
100. Li F, Fang R, Rao L, Meng F, Zhao X. Research progress on exosomes in diagnosis and treatment of cardiovascular diseases. Zhejiang Da Xue Xue Bao Yi Xue Ban 2018;47:320-6. (in Chinese)
101. Dickhout A, Koenen RR. Extracellular vesicles as biomarkers in cardiovascular disease; chances and risks. Front Cardiovasc Med 2018;5:113.
102. Berezin AE, Kremzer A, Berezina T, Martovitskaya Y. The signature of circulating microparticles in heart failure patients with metabolic syndrome. J Circ Biomark 2016;5:1849454416663659.
103. Berezin AE, Kremzer AA, Martovitskaya YV, Samura TA, Berezina TA. The predictive role of circulating microparticles in patients with chronic heart failure. BBA Clin 2014;3:18-24.
104. Berezin AE, Kremzer AA, Berezina TA, Martovitskaya YV. The pattern of circulating microparticles in patients with diabetes mellitus with asymptomatic atherosclerosis. Acta Clin Belg 2016;71:38-45.
105. Bei Y, Chen T, Banciu DD, Cretoiu D, Xiao J. Circulating exosomes in cardiovascular diseases. Adv Exp Med Biol 2017;998:255-69.
106. Zamani P, Fereydouni N, Butler AE, Navashenaq JG, Sahebkar A. The therapeutic and diagnostic role of exosomes in cardiovascular diseases. Trends Cardiovasc Med 2019;29:313-23.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Berezin AE, Berezin AA. Endothelial cell-derived extracellular vesicles in atherosclerosis: the emerging value for diagnosis, risk stratification and prognostication. Vessel Plus 2020;4:15. http://dx.doi.org/10.20517/2574-1209.2020.03
AMA Style
Berezin AE, Berezin AA. Endothelial cell-derived extracellular vesicles in atherosclerosis: the emerging value for diagnosis, risk stratification and prognostication. Vessel Plus. 2020; 4: 15. http://dx.doi.org/10.20517/2574-1209.2020.03
Chicago/Turabian Style
Berezin, Alexander E., Alexander A. Berezin. 2020. "Endothelial cell-derived extracellular vesicles in atherosclerosis: the emerging value for diagnosis, risk stratification and prognostication" Vessel Plus. 4: 15. http://dx.doi.org/10.20517/2574-1209.2020.03
ACS Style
Berezin, AE.; Berezin AA. Endothelial cell-derived extracellular vesicles in atherosclerosis: the emerging value for diagnosis, risk stratification and prognostication. Vessel Plus. 2020, 4, 15. http://dx.doi.org/10.20517/2574-1209.2020.03
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 8
likes
Like This Article 8
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.