
Tumour vasculature targeted anti-cancer therapy

 , ...
, ... Abstract
The tumour vasculature plays an important role in tumour growth and metastasis. Tumour angiogenesis provides more oxygen and nutrients to growing tumour cells, is not as tightly regulated as embryonic angiogenesis, and do not follow any hierarchically ordered pattern. The heterogeneity of the vasculature, high interstitial fluid pressure, poor extravasation due to sluggish blood flow, and larger distances between exchange vessels are potential barriers to the delivery of therapeutic agents to tumours. The prevention of angiogenesis, normalization of tumour vasculature, and enhancement of blood perfusion through the use of monoclonal antibodies against receptor proteins that are overexpressed on proangiogenic tumour cells, and improved, tumour-targeted delivery of therapeutic agents can all be achieved using nanocarriers of appropriate size. Nanomedicines such as polymeric nanoparticles, lipid nanoparticles, micelles, mesoporous silica particles, metal nanoparticles, noisomes, and liposomes have been developed for the delivery of anticancer drugs in combination with antiangiogenic agents. Amongst them, liposomal delivery systems are mostly approved by the FDA for clinical use. In this review, the molecular pathways of tumour angiogenesis, the physiology of tumour vasculature, barriers to tumour-targeted delivery of therapeutic agents, and the different strategies to overcome these barriers are discussed.
Keywords
Angiogenesis
In general, there is an efficient vascular network that supplies blood to normal tissues. The hierarchal architecture and growth of blood vessels are maintained by the balance between pro-apoptotic and anti-apoptotic factors. This balance is controlled by the metabolic demands of the corresponding tissue. Lymphatic channels on the other hand, remove metabolic waste from the interstitium. Thus, the microstructure of the vascular network is capable of supplying adequate oxygen and nutrition to all associated cells[1]. During tumour progression, there is rapid proliferation of tumour tissue. When the tumour reaches a critical size (1~2 mm3), tumour cells located further from the supplying blood vessel become starved of oxygen and nutrients, leading to the impairment of tumour growth by apoptosis or necrosis. In turn, this triggers angiogenesis, the formation of new blood vessels from existing ones[2]. Although tumour angiogenesis provides for tumour growth and a route for metastasis, it is not as tightly regulated as embryonic angiogenesis[2].
Differences between blood vessels of normal and cancer tissues
The growth of tumour blood vessels does not follow any hierarchy. It is typically heterogeneous, tortuous, branches irregularly, and is enlarged circumferentially[3-5]. The endothelial cells, pericytes (multifunctional mural cells that wrap around endothelial cells) and basement membrane of tumour blood vessels are all abnormal[3]: endothelial cells have abnormally loose intracellular associations and focal intercellular openings that are < 2 µm in diameter[6] while their association with multiple layers of the vascular basement membrane is also loose due to high interstitial pressure, leading to hyper-permeable tumour blood vessels and vascular leakage[7].
Tumour blood vessels also have a reduced surface area: volume ratio. The high interstitial pressure, coupled with a reduced surface area, impairs the delivery of oxygen, nutrients, and removal of metabolites. As such, the tumour microenvironment is typically characterized by hypoxia and acidosis which in turn, selects for apoptosis-resistant and metastasis competent tumour cells [Figure 1].
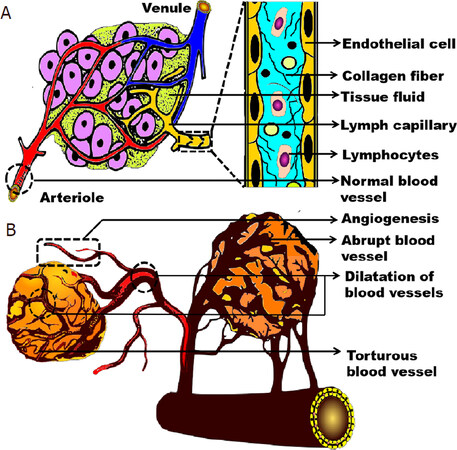
Figure 1. Schematic representation of the physiological differences between normal blood vessels (A) and the tumour vasculature (B)
Cell signalling pathways in hypoxia-induced angiogenesis
Cell signaling pathways in hypoxia-induced angiogenesis is shown in Figure 2. HIF-1α is the founding member of the hypoxia-induced factor (HIF) family[8]. It regulates the genes associated with oxygen deprivation[9]. The HIF activity pathway is regulated by prolyl hydroxylase enzymes (PHD1-3)[10]. PHD acts as an oxygen sensor; in normoxia, PHD hydroxylates the proline residues of HIF-1α. The hydroxylated HIF-1α then binds to the von Hippel-Lindau E3 ubiquitin ligase complex leading to proteasomal degradation of HIF-1α[11,12]. Under hypoxic conditions, oxygen and cofactor 2-oxo-glutarate substrates are depleted[13] and PHD becomes inactivated, resulting in stabilization and intracellular accumulation of HIF-1α. HIF-1α is then translocated into the nucleus to bind with transcriptional factor Arnt (Aryl hydrocarbon nuclear translocator family protein)[14]. Subsequently, a transcriptional complex is formed with p300/CBP which binds to HREs (hypoxia response elements) in the promoters and enhancers of target genes, leading to vasodilatation (for better delivery of oxygen), lowering of oxygen demand and upregulation of proangiogenic factors like fibroblast growth factor (FGF), insulin-like growth factor (IGF), and vascular endothelial growth factor (VEGF)[15]. Vasodilatation is also caused by the upregulation of inducible nitric oxide synthase leading to increased production of nitric oxide and relaxation of vascular smooth muscle cells[16]. Under hypoxic conditions, the demand for oxygen is lowered due to over expression of glucose transporter 1 enzyme (GLUT1). GLUT1 improves the uptake of glucose[17] and induces glycolytic enzymes such as phosphoglycerate kinase[18]. In turn, phosphoglycerate kinase is regulated by aldolase A and HIF-α. Aldolase A helps in better utilization of glycolysis, tumour epithelium mesenchymal cell proliferation[19] and upregulation of pyruvate dehydrogenase kinase (PKD1) which inhibits mitochondrial respiration[20]. HIF-1α helps in cancer cell proliferation[21] by regulating the expression of a number of proangiogenic genes like VEGF, Ang-1, Tie 2, platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), monocyte chemoattractant protein-1 (MCP-1), IGF and epidermal growth factor (EDGF). These HIF regulated factors bind to corresponding receptors on the cell membranes of pericytes and increase vascular permeability, endothelial cell proliferation, sprouting, migration, adhesion, and tube formation. The angiogenic factors, their corresponding receptors, and functions are shown in Table 1. Vascular permeability is increased due to overexpression of VEGF[22-25]. In endothelial cells and pericytes, Ang-1 (angiopoietin-1) is induced by hypoxia. It is a Tie-2 receptor agonist which recruits pericytes to mature vessels and promotes tumour angiogenesis[22]. Despite active angiogenesis, the tumour microenvironments still have hypoxic domains that lead to sustained stabilization of HIF-α. HIF-α then promotes cap-dependent translation of selective mRNAs for angiogenesis through up-regulation of translational factor eIF4E1. In contrast, 4E-BP1 is a translation initiation repressor that sequesters eIF4E1 and is thus a tumour supressor protein. The activity of translational factor eIF4E1 is also controlled by pathways such as Ras and PI3K/AKT. These pathways act by inhibiting 4E-BP1 and increasing the expression of eIF4E1.
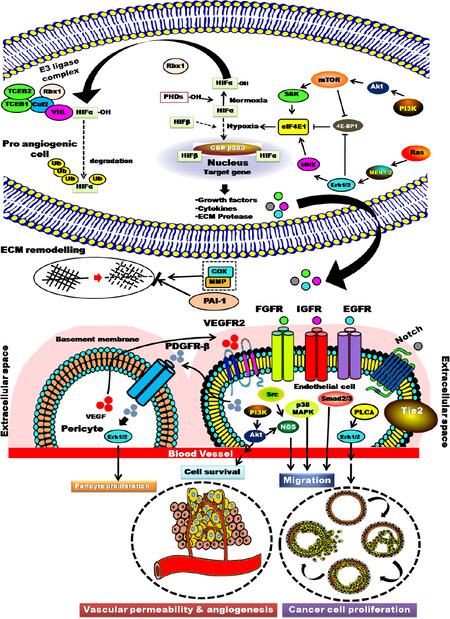
Figure 2. Cell signalling pathways of hypoxia-induced tumour angiogenesis. MNK: mitogen-activated protein kinase interacting protein kinases; EGFR: endothelial growth factor; VEGFR2: vascular endothelial growth factor receptor type 2; PDGFR: platelet derived growth factor receptor; VEGF: vascular endothelial growth factor; ECM: extracellular matrix; MMP: matrix metalloproteinase; mTOR: mammalian target of rapamycin; TCEB: transcription elongation factor B; FGFR: fibroblast growth factor receptor; IGFR: insulin-like growth factor receptor
List of angiogenic factors, corresponding receptors, and functions
| Antigenic molecules | Receptors | Functions | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Initiation of angiogenesis | Neovessel formation | Adaptation to tissue needs | Maturation | ||||||||
| Enhancement of vascular permeability | Detachment of pericytes | Degradation of basement membrane | Endothelial cell proliferation and migration | Pericyte proliferation and migration | Regression of neovessels due to lack of flow or presence of growth factors | Attachment of pericytes | Deposition of basement membrane | Endothelial assembly and lumen acquisition | Vessel maintenance | ||
| VEGF | VEGFR1 (Flt1) VEGFR2 (Kdr) | √ | √ | √ | √ | √ | |||||
| Ang-2 | Tie2 | √ | √ | √ | |||||||
| FGF | FGFR | √ | √ | ||||||||
| PDGFB | PDGFR | √ | √ | √ | √ | ||||||
| PLGF | VEGFR1 (Flt1) | √ | |||||||||
| THBS 1 | CD36, CD47, Integrins | √ | |||||||||
| Integrins | Extracellular matrix | √ | √ | ||||||||
| SDF1 | CXCR4 | √ | |||||||||
| DLL1-4 | Notch | √ | |||||||||
| SCF | cKit | √ | |||||||||
| Interleukins | Interleukin receptors | √ | |||||||||
| Ang-1 | Tie2 | √ | √ | √ | √ | ||||||
The inducible enzyme cyclooxygenase-2 (COX-2) is also an important mediator of angiogenesis and tumor growth. It induces matrix metalloproteinases that have traditionally been associated with the degradation and turnover of most of the components of the extracellular matrix (ECM). Plasminogen activator inhibitor type 1 (PAI-1) though has the opposite effect of remodeling the ECM by regulating plasmin.
Barriers to targeted delivery of therapeutic agents to tumour
Spatial and temporal heterogeneities in blood supply
Vascular morphology and blood flow rate govern the movement of blood-borne particles through tumour vasculature. Depending on the tumour type, location and growth rate, the architecture of the tumour vasculature may vary[26]. Blood vessel distribution throughout the tumour mass is also not uniform and each region may have either peripheral or central vascularization. In other words, the central portion of some regions is well perfused whereas the periphery may have better perfusion elsewhere.
Microscopically, the tumour vasculature is highly heterogeneous. They are characterized by dilated, secular and tortuous blood vessels having tri-furcations, self-loops, and sprouts. The endothelial cell lining may even be absent. Blood flow is also chaotic and lacks a definite route between the arterial and venous systems. Therefore, in general, necrotic foci develop in a growing tumour. In turn, this decreases the average rate of perfusion.
Based on the rate of perfusion, there may be four regions in a tumour[26]: (1) an avascular, necrotic region; (2) semi-necrotic region; (3) stabilized, microcirculation region; and (4) advancing front.
Regions I and II have a low blood flow rate whereas in regions III and IV, flow is more variable but still higher than that of surrounding normal host tissue. With tumour growth, the widths of regions I and II increase while that of III and IV remain unchanged, resulting in variation in vascular morphology at both the macroscopic and microscopic levels. The resulting spatial and temporal heterogeneities in blood supply is thus responsible for non-uniform distribution of the therapeutic agent. Generally, the average uptake of a therapeutic agent decreases with an increase in tumour mass.
Poor extravasation and high interstitial fluid pressure limit transport across the microvascular wall
Diffusion and convection are the main mechanisms behind the transport of drug molecules across the vascular wall. The concentration gradient of the therapeutic agent across the plasma (Cp) and interstitial fluid (Ci) is the driving force for the diffusion process. This mass transfer process is proportional to the surface area; the proportionality constant is known as vascular permeability P (cm/s). Transfer of therapeutic agents by convection is associated with the leakage of plasma/fluid across the vascular wall due to differences in hydrostatic pressure of fluid in the blood vessel and interstitial space. The associated experimental constant is known as hydraulic conductivity, Lp (cm/mmHg-s). Similarly, the convection process is also proportional to the osmotic pressure difference between the blood vessel and the interstitial space[27]. This proportionality constant is known as the osmotic reflection coefficient (σ). These three experimental constants (P, Lp, and σ) are used to describe the extent of transport of plasma content across tumour vessels. Tumour vessels have relatively high P and Lp values[28,29] as they have wide endothelial junctions, a large number of fenestrae and trans-endothelial channels, discontinuous or absent basement membrane and significant spatial heterogeneities[30,31]. Although these physiological characteristics increase vascular permeability, tumours also have poor extravasation, which is a significant barrier to the delivery of therapeutic agents. This can be explained as follows: tumour vessels have sluggish blood flow. The hydrostatic fluid pressure in the blood vessel (Pv) is less than that of fluid in the interstitial space (Pi). Of note, the Pi in animal/human tumours is even higher than that of normal tissue[32]. Furthermore, it has been reported that Pi increases with the growth of a tumour. This is mainly due to high vascular permeability and poor, impaired lymphatic drainage[32-35]. Both tumour hyperplasia around a blood vessel and increased production of extracellular matrix components contribute to high interstitial fluid pressure (IFP). In normal tissue, IFP is 0 mmHg but in tumour blood vessel, the IFP varies from 10-40 mmHg[36]. The IFP is elevated throughout the mass of a tumour except at the periphery, where it becomes equal to normal physiological values. Therefore, intratumoral fluid may extravasate from the periphery of a tumour, resulting in non-delivery of a therapeutic agent. In different animal and human tumour models, it was found that 1%-14% of plasma entering the tumour leaked into the periphery[28,37,38]. Again, the tumour interstitial space has a higher concentration of endogenous plasma protein, leading to higher interstitial osmotic pressure. Thus, the transfer of therapeutic agents by diffusion is further limited.
Resistance to transport through the interstitial space and distribution into the tumour microenvironment
Diffusion and convection are the main mechanisms behind the movement of therapeutic agents that have extravasated into the interstitial space[39]. The concentration gradient is the driving force behind diffusion whereas fluid velocity determines the convection process. The interstitial diffusion coefficient (D) and hydraulic conductivity (K)[32] are the experimental constants used for quantitative measurements of therapeutic agent distribution in the interstitial space. The interstitial space of a tumour is located at the TME (tumour microenvironment) and composed largely of a collagen and elastic fibre network, filled with a hydrophilic gel made up of interstitial fluid and macromolecular constituents[40]. Its structural integrity is maintained by collagen and elastin whereas resistance to transport is provided by macromolecular constituents such as glycosaminoglycans and proteoglycans[40,41]. Compared to normal tissues, tumours have a higher collagen content but lower concentrations of hyaluronate and proteoglycans[32] due to increased activity of lytic enzymes such as hyaluronidase in the tumour interstitial space. Thus, the tumour interstitial space should provide lower resistance to the distribution of therapeutic agents, suggesting larger values of D and K. Paradoxically however, therapeutic agents are not distributed homogeneously in tumours. This can be explained as follows: the time constant for a molecule with diffusion coefficient, D is proportional to the diffusion path length, raised to a power of two. Therefore, if the diffusion path length is doubled, the required time will be increased by four times. In solid tumours, the exchange vessels are at a large distance apart (~200 µm)[42,43]. Therapeutic agents will need a prolonged transit time for homogenous distribution. High interstitial pressures also slow down the distribution process. Thus, low molecular weight (Mr < 1000 Da) anticancer drugs do not accumulate in the tumour because of their small size and hence, rapid clearance[44]. The drug distribution process in a tumour may be further limited by the high affinity of the drug molecule for proteins present in interstitial fluid.
Growth induced solid stress
A tumour mass consists of proliferating cancer cells and stromal cells (i.e., fibroblasts, immune, and perivascular cells)[45]. It is supplied by a dense ECM, and a tortuous and chaotic network of blood vessels[45]. During tumour growth, there is rapid proliferation of cancer cells in a limited space resulting in the generation of mechanical forces from different structural components such as cancer cells, various host cells, and the ECM. Thus, there is also a growth induced solid stress, which commonly ranges from 10 to 142 mmHg[46], that can deform the vascular and lymphatic structures and cause limited perfusion and hypoxia throughout tumour tissue. This creates a barrier to the penetration of therapeutic agents[47] which restricts their flow to cells within the perivascular space, such that resistant cells in hypoxic regions are missed[45]. Shear stress can also induce vascular endothelial growth factor receptor type 2 (VEGFR2) expression and ligand-independent phosphorylation. This causes activation of MAPK, PI3K, and Akt signalling pathways that are involved in promoting angiogenesis[46]. Additionally, there is VEGFR2 membrane clustering and downstream signalling. Recently VEGFR3 has also been found to be a part of this mechanosensory complex. Depletion of VEGFR2 or VEGFR3 thus causes significant reduction in endothelial cell response to mechanical stress[46].
Specific integrins can also contribute to tumour angiogenesis and tumour progression[46]. In endothelial cells, VEGF upregulate the expression of α1β1 and α2β1 integrins. The α5β1, αvβ3 and αvβ5 integrins are also expressed in angiogenic vasculature to facilitate the growth and survival of newly forming vessels[46].
Therefore, the general strategy to overcome the barriers to vascular and tumour tissue permeability is functionalization of the surface of nanoparticles with tissue and cell-penetrating peptides, such as the iRGD[48]. It interacts with αν integrins on the endothelium and stimulates proteolytic cleavage. The released CendR peptide subsequently binds with neuropilin-1[45] to ensure the homing of and penetration of tumour tissue by nanoparticles.
Targeted delivery of therapeutic agents by exploiting tumour vasculature
A therapeutic agent is delivered to the target tissue via supplying arterioles to that particular tissue. As discussed in the previous sections, there are a number of barriers that hinder the distribution process of therapeutic agents in the tumour. First, the tumour vasculature is highly heterogeneous in distribution. Unlike the tight endothelium of normal blood vessels, the vascular endothelium in tumour microvessels is discontinuous and leaky. Elevated levels of growth factors such as VEGF and bFGF cause vasodilatation and enhancement of vascular permeability. Therefore, the gap sizes between endothelial cells can range from 100 to 780 nm, depending on the anatomic location of the tumour[49]. As such, low molecular weight anticancer drugs (Mr < 1000 Da) can easily enter the tumour microenvironment but at the same time, they can also be easily removed because of their small size. Consequently, when delivered as an aqueous solution, small-molecule chemotherapeutic agents like paclitaxel[50], gemcitabine[51], cisplatin[52], etc. do not accumulate in the tumour at the desired concentration for an adequate duration. These potent anticancer drugs undergo unwanted bio-distribution, leading to unfavourable pharmacokinetics characterized by a large volume of distribution, high renal clearance and short half-life[53]. Furthermore, these cytotoxic agents can cause severe dose-dependent side effects such as myelosuppression, neurotoxicity, mucositis, nausea, vomiting, and alopecia that may become fatal for patients[54], or even, the development of drug resistance and relapse of cancer[55].
This problem can potentially be solved by delivering anticancer drugs encapsulated within nanoparticles[56,57] or as drugs conjugated to the nanoparticle’s surface[58-61]. Due to their size range, nanoparticles are inherently able to permeate through leaky tumour microvessels but impaired lymphatic drainage of the solid tumour, together with a higher interstitial fluid pressure, hinders clearance of nanoparticles from the TME. Thus, retention of anticancer drugs is enhanced when delivered as nanomedicine. This mechanism of passively targeting a solid tumour is known as the enhanced permeation and retention (EPR) effect, which was first described by Matsumura and Maeda[62] in 1986.
The size of the tumour, degree of tumour vascularization, and angiogenesis are the main factors affecting EPR[63-65]. Thus, the stage of the disease is critical for drug targeting using the EPR effect[66]. Another factor is the challenge for the chosen delivery system to penetrate deep into tumour tissue due to the high interstitial fluid pressure at the centre of a tumour[67]. This results in initial tumour regression, followed eventually by recurrence from residual cells in the non-accessible regions of the tumour[68]. Therefore, the drug delivery system needs to be optimized for deep tumour penetration[69-71]. This can be achieved by (1) enhancing blood perfusion to a tumour; (2) modulating the structure of tumour vasculature; and (3) destroying the mass of cancer cells to increase passage of nanoparticles.
Enhancing blood perfusion to a tumour
As discussed earlier, tumour blood vessels have sluggish blood flow. The hydrostatic fluid pressure in a blood vessel (Pv) is less than that of fluid in the interstitial space (Pi). This limits the distribution of therapeutic agents in the TME. Therefore, an increased rate of blood flow in tumour vessels will enhance the distribution of nanoparticles in the TME because of higher extravasation. Strategically there are two ways to increase the rate of blood flow in tumour vessels. First, vasoconstrictors such as angiotensin can be parenterally administered[72]. This will constrict normal blood vessels but not tumour blood vessels which will remain unaffected because of their impaired muscular structure. As a result, more blood will be delivered to tumour blood vessels. Second, vasodilators like NO and CO should be delivered directly to tumour blood vessels without affecting blood vessels of normal tissue[73].
In experimental rats with subcutaneously transplanted AH109A solid tumours, Suzuki et al.[74] found a 5.7 fold enhancement of blood flow in the tumour after intravenous administration of angiotensin II. This enhanced the chemotherapeutic effect of mitomycin C on the main tumour and metastatic foci in lymph nodes. Nagamitsu et al.[72] then successfully treated patients with SMANCS (neocarzinostatin, the anti-tumour antibiotics conjugated with a hydrophobic copolymer of styrene) under angiotensin induced hypertensive states. The induction of hypertension at ~15-30 mm Hg higher than normal blood pressure for 15-20 min resulted in remarkably enhanced and passively targeted delivery of neocarzinostatin to the tumour. This resulted in faster reduction of tumour size with the least toxicity to normal tissue.
Many research groups have developed nano-medicines that induce tumour-specific vasodilatation by releasing mediators such as NO[75,76] and CO[73]in situ. This helped in the accumulation of nanoparticles within the TME. Tahara et al.[77] incorporated NONOate, a typical NO donor, into PEGylated liposomes. Its retention in blood was similar to that of empty PEGylated liposomes but its accumulation within the tumour was doubled. Due to successful augmentation of the EPR effect, this liposome could be a potential vehicle for the targeted delivery of potent chemotherapeutic agents.
Wei et al.[78] then developed tumour vascular-targeted multifunctional hybrid polymeric micelles for the targeted delivery of doxorubicin [Figure 3]. Poly (d,l-lactide) (PLA) and poly (ε-caprolactone) (PCL) formed the inner core to encapsulate doxorubicin. The poly (ethylene glycol) (PEG) was linked to PLA with disulphide linkages to form the outer surface of the particle. Copper ion-chelated porphyrin (PpIX-Cu) was then added to the end of the PEG segment, providing a catalytic function to decompose endogenous NO donors like S-nitroso-glutathione (GSNO), S-nitrosocysteine, and S-nitrosoalbumin. Since these endogenous NO donors are also present in human plasma and all tissue fluid, 2-propionic-3-methyl-maleic anhydride (CDM)-modified methoxy polyethylene glycol (mPEG) (mPEG-CDM) was linked to the PpIX-Cu component as a pH-sensitive protective layer, in order to mask the positive charges of the micelles and avoid copper ion-catalysed NO production in the general circulation. Copper catalysed NO production occured only in mildly acidic (pH 6.5) tumour tissue. Furthermore, cRGD grafted PCL-PEG-cRGD (PCE-cRGD) copolymer was added during the synthesis of micelles. The grafted cRGD peptide then effectively targeted the tumour vasculature and tumour cells, on which αvβ3 integrin is overexpressed. Once taken up by the cancer cell, doxorubicin was immediately released due to the high cytoplasmic level of GSH. Thus, this complex hybrid polymeric micelle structure was very effective in treating tumours in an animal model.
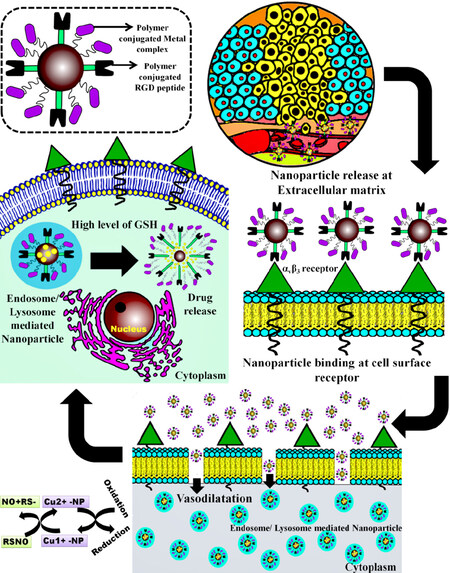
Figure 3. Schematic representation of NO generating tumour vasculature targeted drug delivery systems. Copper ion-chelated porphyrin triggers tumour vasculature specific release of NO causing local vasodilation, whereas RGD peptide causes αvβ3 mediated tumour cell-specific nanoparticle uptake. The drug is released specifically within the cancer cells where the cytoplasmic levels of GSH is higher than normal cells. NO: nitric oxide; GSH: glutathione; RSNO: S-Nitroso alkane NP: nanoparticle; RGD: arginylglycylaspartic acid
Fang et al.[79] reported augmentation of the EPR effect and efficacy of anticancer nanomedicine by CO generating agents. Haem oxygenase (HO) catalyses the degradation of haem to produce CO which causes vasodilatation similar to NO[80-82]. Pegylated hemin is the HO inducer whereas tricarbonyl-di-chloro-ruthenium (II) dimer (CORM2) is the CO-releasing molecule[79]. The authors showed that in tumour-bearing mice, the accumulation of intravenously administered Evans blue-albumin complex (a macromolecule) in a tumour can be enhanced by the intradermal injection of recombinant haem oxygenase-1, intra-tumoral injection of tricarbonyl-dichloro-ruthenium (II) dimer (CORM2) and intravenous administration of PEGylated hemin. Thus CO plays a significant role in tumour uptake of macromolecular drugs by EPR[83]. They have also developed polymeric micelles of CORM2 copolymer and styrene maleic acid. It had a prolonged plasma half-life and was able to maintain a sustained release of CO. They used it for photodynamic therapy with pyropheophorbide-a[79].
Modulating the structure of tumour vasculature
The balance between pro-angiogenic (e.g., VEGF, PDGFB, IGF, PDGFRB, FGF-2, and TIE2) and anti-angiogenic factors (e.g., thrombospondin-1, angiostatin and endostatin) is responsible for the formation of normal tissue vasculature. This balance tips in favour of overexpression of pro-angiogenic factors in pathological conditions such as the progression of solid tumours[84]. The purpose of such is to meet the high demand for oxygen and nutrients of tumour cells. Therefore, restoring this balance of factors may restore tumour vasculature back to normal. This process involves the inhibition of pro-angiogenic factors at a different level of their cell signalling pathways [Figure 2], which will reduce the diameter of tumour microvessels, prune immature vasculature, increase vasculature maturity with higher pericyte coverage, reduce tortuosity of microvessels, and decrease IFP. Although normalization of tumour vasculature is the rationale for inhibition of tumour growth[85], it is not effective enough alone in clinical settings. Instead, it has been found in clinical trials that combinations of radiotherapy or chemotherapy together with anti-angiogenic agents are very effective[84,86]. Ionizing radiation generates ROS that leads to DNA damage and cell death. Since the presence of oxygen helps in the generation of ROS, a well-vascularized and perfused tumour tissue would be more susceptible to radiotherapy[86]. It has also been shown that under low-dose irradiation, cancer cells are induced to express proangiogenic factors (e.g., VEGF, PIGF) at a level sufficient to stimulate endothelial cell migration and sprouting. This is known as the vascular rebound effect[87], which can be overcome by combining anti-angiogenic agents with radiotherapy. In one clinical trial on advanced pancreatic cancer patients, a combination of optimal dosages of bevacizumab, capecitabine and radiotherapy was found to be very effective[88]. In another clinical study with rectal cancer patients, promising results were reported when radiotherapy was combined with bevacizumab, capecitabine, and oxaliplatin[89]. In cases of chemotherapy used in combination with anti-angiogenic agents, normalization of tumour vessels will not only reduce vascular permeability but at the same time, enhance the trans-capillary pressure gradient (due to lowering of IFP), resulting in better distribution of small molecule anticancer drugs and nanoparticles (< 60 nm) into the TME[84].
Strategically, one may either block the pathways for synthesis of pro-angiogenic factors and their target receptor proteins, or neutralize the effects of these factors by inhibiting the corresponding target receptors with monoclonal antibodies. Such angiogenesis inhibitors can either target endothelial cells of the growing vasculature (known as direct inhibitors) or tumour cells and tumour-associated stromal cells (indirect inhibitors). Direct inhibitors like angiostatin[90], endostatin[91], arrestin[92], canstatin[93] and tumastatin[94,95] bind with integrin receptor to prevent the proliferation and migration of endothelial cells in response to different pro-angiogenic factors. Indirect inhibitors prevent the expression of pro-angiogenic proteins (e.g., VEGF) expressed by tumour cells or block the expression of corresponding endothelial cell receptors (VEGFR). Many angiogenesis inhibitors have been approved by the FDA for cancer therapy including thalidomide[96], bevacizumab[97], pazopanib[98] and everolimus[99] amongst others. There are also many candidate anti-angiogenic drug molecules such as siRNA, shRNA, VEGF aptamer, KPQPRPLS-peptide currently under study.
Different types of nanomedicines such as polymeric nanoparticles, lipid nanoparticles, micelles, mesoporous silica particles, metal nanoparticles, noisomes, and liposomes have been developed for the delivery of anticancer drugs. Amongst them, liposomal delivery systems are mostly approved by the FDA for clinical use.
Therapeutic nucleic acids like small interfering RNA (siRNA) and short hairpin RNA (shRNA) are negatively charged and thus, frequently delivered with liposomes made up of cationic phospholipids. Cai et al.[100] developed Bio-reducible fluorinated peptide dendrimers for efficient and safe delivery of VEGF siRNA. It improved physiological stability, serum resistance; promoted intratumoral enrichment, cellular internalization, as well as facilitated endosomal/lysosomal escape and reduction-triggered cytoplasm siRNA release. It had found to have excellent VEGF gene silencing efficacy (~65%) and a strong ability to inhibit HeLa cell proliferation. Upon intratumoral injection in mice with HeLa tumor xenografts, it significantly retarded tumour growth. Yang et al.[101] developed strategy for co-delivery of VEGF siRNA and docetaxel. This dual peptide modified liposome binds specifically to glioma cells, undergoes specific receptor-mediated endocytosis and deep tissue penetration. Once within target cells, the siRNA silences the VEGF gene to inhibit angiogenesis while docetaxel kills tumour cells.
Chen et al.[102] studied the effect of silencing the VEGF gene using siRNA for the treatment of breast cancer (MCF7 xenograft model) with doxorubicin. They prepared calcium phosphate/siRNA nanoparticles and further encapsulated it in a liposome. The liposome was injected intratumorally while doxorubicin was administered intraperitoneally. This combination therapy resulted in 91% tumour inhibition using only 60% of the standard dose of doxorubicin. In a more recent study, Zheng et al.[103] utilized mesoporous silica nanocarriers (148.5 nm) for the co-delivery of sorafenib (a multikinase inhibitor) and VEGF targeted siRNA to treat hepatocellular carcinoma. The particles were further coated with lactobionic acid to target asialoglycoprotein receptors that are overexpressed on cancer cells. Taking one step further, Shen et al.[104] co-delivered sorafenib and survivin shRNA with nano-complexes to reverse multidrug resistance in human hepatocellular carcinoma. Survivin is an angiogenesis promoting agent. Suppression of survivin with shRNA thus resulted in the reversal of drug resistance and promoted sensitization to sorafenib treatment, leading to cell cycle arrest and apoptosis.
While positively charged liposomes are best suited for the delivery of negatively charged RNA molecules, they undergo nonspecific electrostatic adsorption with blood components and are quickly recognized by the immune system, leading to rapid clearance from the blood by the reticuloendothelial system (RES). This limitation can be overcome by coating the positively charged liposomes with negatively charged anionic polymers, which would then prolong circulation of the nanoparticles in blood and enhance the accumulation of nanoparticles within the tumour due to the EPR effect. In a recent study, VEGF siRNA and etoposide were loaded in a cationic liposome that was further coated with PEGylated histidine-grafted-chitosan-lipoic acid (PHCL), a pH triggered charge-controllable and redox responsive polymer [Figure 4][105].
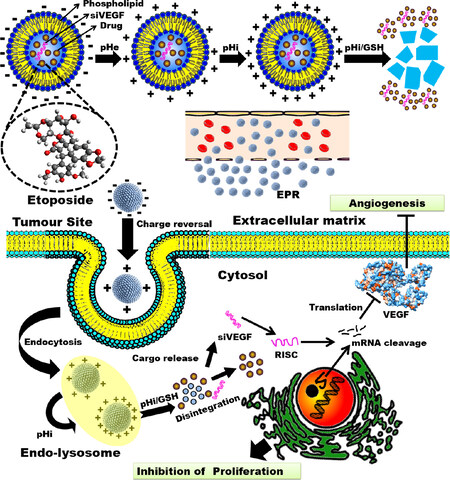
Figure 4. Schematic representation of using multifunctional nanoparticles for co-delivery of VEGF siRNA and etoposide (an anticancer drug) for enhanced anti-angiogenesis and anti-proliferation activity. RISC: siRNA induced silencing complex; VEGF: vascular endothelial growth factor; GSH: glutathione; EPR: enhanced permeation & retention
In the TME, at low pH (6.5), protonation of the imidazole group in the histidine segment of PHCL causes a reversal of nanoparticle charge from negative to positive, leading to deep tumour penetration and enhancement of internalization of nanoparticles. The positive charge is further enhanced in the lower pH of endo-lysosomes, where the disulphide bond of the lipoic acid segment in PHCL-liposomes undergo GSH induced redox-activated breakage, leading to the release of cargo within the liposome [Figure 4].
The antiangiogenic agent bevacizumab is a humanized monoclonal antibody that inhibits tumour growth and metastasis. When combined with a cytotoxic anticancer agent such as paclitaxel, therapeutic efficacy was significantly improved because of the targeted accumulation of paclitaxel within tumours[106]. In a preclinical study using the MX-1 human breast cancer xenograft model, different doses of paclitaxel were administered in combination with 5 mg/kg bevacizumab. 30 mg/kg paclitaxel in combination with bevacizumab was as effective as 100 mg/kg single dose of paclitaxel in inhibiting the growth of a tumour. This observation can be attributed to treatment with bevacizumab, which significantly enhances the effective concentration of paclitaxel within the tumour.
Gold nanoparticles have also been used for the targeted delivery of anti-angiogenic agents, either alone or in combination with an anticancer drug. Bartczak et al.[107] synthesized gold nanoparticles of ~15 nm and capped them with mono-carboxy (1-Mercaptoundec-11-yl) hexa (ethylene glycol). These particles were then further functionalized through surface coating with a peptide (KATWLPPR) that specifically binds to neuropilin-1 receptor to inhibit angiogenesis. In an in vitro study using human endothelial cells, it was found that this peptide coated gold nanosphere could block capillary formation by endothelial cells without causing toxicity. Patra et al.[108] then used gold nanoparticles for targeted co-delivery of cetuximab and gemcitabine. Cetuximab has been approved for the treatment of EGFR positive colorectal cancer whereas gemcitabine is used for pancreatic carcinoma. “2 in 1” nanoconjugates containing both cetuximab and gemcitabine on a single gold nanoparticle core were synthesized. Physically, this was more stable than a gold nanoparticle-containing either of the agents. This nanoconjugate could target metastatic EGFR expressing cells and inhibited 80% tumour growth and was significantly better than all other non-targeted groups.
EGFR tyrosine kinase inhibitors like cetuximab, lapatinib, afatinib, gefitinib, erlotinib, fedratinib are well studied for anticancer therapy when used in combination with different chemotherapeutic agents including doxorubicin, gemcitabine, paclitaxel, and carboplatin. They help in the normalization of tumour vasculature and sensitize tumour cells to cytotoxic drugs. Additionally, monoclonal antibodies such as cetuximab have been used as a targeting agent. Lin et al.[109] conjugated both paclitaxel and cetuximab on the surface of carbon nano-diamond particles of 3-5 nm diameter. This was found to enhance the mitotic catastrophe and tumour inhibition in the drug resistance of colorectal carcinoma in vitro and in vivo. Among the other inhibitors, lapatinib also inhibits human epidermal growth factor receptor 2 (HER2) tyrosine kinases and ATP-binding cassette transporters, thereby sensitizing multidrug-resistant (MDR) cancer cells to chemotherapeutic agents. Lapatinib was clinically approved by the US FDA in 2007 for anticancer therapy. There have been many studies since where lapatinib has been used in combination with paclitaxel, and liposomes and polymeric micelles used as drug delivery vehicles. Li et al.[110] developed stealth polymeric micelles using an amphiphilic diblock copolymer named poly (ethylene glycol) -block-poly (2-methyl-2-carboxyl-propylene carbonate-graft-dodecanol) which formed a core-shell structure by self-assembly. Hydrophobic molecules like paclitaxel, lapatinib are loaded into the hydrophobic core while the hydrophilic shell of PEG prevents their aggregation, restricts plasma protein adsorption, prevents recognition by the RES, and minimizes rapid elimination from the bloodstream. This ~60 nm particle successfully overcame multidrug resistance in an athymic nude mouse xenograft model established with DU145-TXT MDR prostate cancer cells. The strategies of tumour-targeted drug delivery exploiting tumour vasculature aresummarised in Table 2. The FDA-approved anti-angiogenic agents for the treatment of cancer is summarized in Table 3.
Strategies of tumour-targeted drug delivery exploiting tumour vasculature
| Proangiogenic factor | Antiangiogenic agent | Anti-cancer drug | Formulation/delivery system | Mechanism of action | In vivo/ex vivo/clinical study | Year of study | Ref. |
|---|---|---|---|---|---|---|---|
| VEGF | siRNA | Not applicable | Liposome with two peptides (Angiopep and tLyP-1) attached on the surface | Angiopep ligand helps in brain tumour targeting, tLyP-1 ensures tumour penetration. siRNAs inhibit VEGF production | In vivo: nude mice bearing U87 MG glioblastoma | 2014 | [111] |
| Not applicable | cis-di-ammine-di-nitro-platinum (II) | Anti-VEGF mAb and anti-VEGFR2 mAb were attached on the liposome surface | The mAb targets the liposome to tumour cells. Cis-di-ammine-di-nitro-platinum (II) kills cancer cells | Ex vivo: glioma C6 and U-87 MG cells
In vivo: intracranial C6 glioma rat model using female Wister rat | 2016 | [112] | |
| Sorafenib and Cy3-siRNA | Not applicable | pH-sensitive carboxymethyl chitosan-modified liposomes | Inhibition of angiogenesis due to downregulation of VEGF | Ex vivo: HepG2 cell
In vivo: H22 tumour-bearing mice | 2019 | [113] | |
| Not applicable | DOX | DOX-loaded Amino-triphenyl dicarboxylate-bridged Zr4+ metal-organic framework
Nanoparticles gated with a duplex nucleic acid including an anti-VEGF aptamer in a caged configuration | VEGF overexpressed by cancer cells provides the mechanism to unlock the gate via the formation of the VEGF-aptamer complexes and the separation of the gating duplex. The released DOX kills the cancer cells | Ex vivo: MDA-MB-231 breast cancer cell line | 2018 | [114] | |
| siRNA | DOX HCl | Polycation liposome-encapsulated calcium phosphate nanoparticle | siRNA silences the expression of VEGF. DOX kills cancer cells | Ex vivo: MCG-7 cell line
In vivo: MCF-7 xenograft tumour model in nude mice | 2017 | [115] | |
| Gambogic acid | Gambogic acid | PEGylated liposomes | Gambogic acid has both antiangiogenic and cytotoxic activity | Ex-vivo: MDA-MB-231 cells
In vivo: MDA-MB-231 orthotopic xenograft model | 2016 | [116] | |
| siRNA | Docetaxel | Liposome with two peptides (Angiopep and tLyP-1) attached on the surface | Angiopep ligand helps in brain tumour targeting, tLyP-1 ensures tumour penetration. siRNA inhibits VEGF production. Docetaxel kills cancer cells | Ex vivo: human glioblastoma cells (U87 MG) and murine BMVEC
In vivo: male BALB/c nude mice with U87 MG tumours | 2014 | [117] | |
| siRNA | Etoposide | Cationic liposomes coated with PEGylated histidine-grafted chitosan-lipoic acid | siRNA silence VEGF gene. Etoposide kills cancer cells | Ex-vivo: A549-Luc In vivo: nude mice bearing orthotopic A549-Luc tumour | 2019 | [105] | |
| Bevacizumab | Paclitaxel | Bevacizumab diluted with saline, paclitaxel dissolved in 1:1 mixture of cremophor el and ethanol solution | Inhibiting the binding of VEGF to its cell surface receptors with the anti-tubulin agent | In vivo: MX-1 human breast cancer xenograft model and A549 xenograft model | 2010 | [118] | |
| siRNA | Sorafenib | Lactobionic acid conjugated mesoporous silica nanoparticle | siRNA inhibits VEGF expression. Sorafenib has antiangiogenic and cytotoxic effects | Ex vivo: asialoglycoprotein receptor overexpressing hepatocellular carcinoma (HepG2, Huh7) cells | 2018 | [103] | |
| shRNA (Survivin) | Sorafenib | Pluronic P85- Poly-ethyleneimine/D-α-tocopheryl-PEG 1000 succinate nanocomplexes (nanomicelle) | shRNA inhibits VEGF expression. Sorafenib has antiangiogenic and cytotoxic effects | Ex vivo: multidrug resistance hepatocellular carcinoma cells (BEL-7402)
In vivo: xenograft model in nude mice | 2014 | [104] | |
| Vatalanib | Not applicable | Oral tablet | Vatalanib is an angiogenesis inhibitor. It inhibits the tyrosine kinase domains VEGFR, PDGFR, and c-KIT | Clinical (Phase II): patients with metastatic pancreatic adenocarcinoma who failed first-line treatment with gemcitabine | 2014 | [119] | |
| Sorafenib | Paclitaxel | Hyaluronic acid conjugated D-α-tocopheryl polyethylene glycol 1000 succinate and polylysine-deoxycholic acid copolymer co-modified cationic liposome | Sorafenib is an angiogenesis inhibitor. It also inhibits cancer cell proliferation (by inhibiting RAF/MEK/ERK signalling pathways). Paclitaxel arrests cancer cells at G2/M phase | Ex vivo: multi-drug resistant MCF7 breast cancer cell line
In vivo: xenograft model using BALB/c nude mice | 2019 | [120] | |
| Sunitinib | Near-Infrared dye-IR780 | Liposome | Laser activated release of sunitinib inhibits tyrosine kinase associated with VEGF and PDGF receptors, whereas IR780 dye kills cancer cells by hyperthermia | Ex-vivo: 4T1 cell line
In vivo: BALB/c mice bearing 4T1 tumours | 2018 | [121] | |
| Sunitinib | Paclitaxel | Paclitaxel loaded pH-responsive micelle was coated with β-cyclodextrin via MMP-2 sensitive peptide that was cleavable in the tumour matrix. Sunitinib was loaded in this cyclodextrin layer | Drugs were released at the tumour microenvironment (low pH, presence of MMP). Sunitinib inhibits angiogenesis and paclitaxel arrests cancer cells at the G2/M phase | Ex vivo: C6 glioma cell
In vivo: C6 tumour bearing nude mice | 2019 | [122] | |
| KATWLPPR peptide | Gold nanoparticle | Gold NP capped with monocarboxy (1-mercaptoundec-11-yl) hexa (ethylene glycol) | Gold nanoparticle delivers the peptide within the cell, where it predominately binds to neuropilin-1 receptor and inhibits angiogenesis | Ex vivo: human breast cancer cell lines (MCF-7 and MDA-MB-231) | 2013 | [107] | |
| FGF | FGF1 (recombinant ligand for all FGFRs) | Gold nanoparticle (AuNP) | FGF1 conjugated gold nanoparticle | FGF1 helps in the targeted delivery of AuNP to FGFR positive cells to cause NIR induced photothermal destruction of cancer cells | Ex vivo: BJ cells and mouse fibroblast (NIH 3T3) cells | 2012 | [123] |
| Epidermal growth factor | Cetuximab | Paclitaxel | Cetuximab conjugated paclitaxel loaded nanodiamond | Cetuximab helps in cancer cell-targeted delivery of paclitaxel that arrests cells at G2/M phase | Ex-vivo: human colorectal cell line (HCT116, SW620, and RKO)
In vivo: a special strain of Balb/C mice bearing subcutaneous tumour | 2017 | [109] |
| Cetuximab | Gemcitabine | “2 in 1” nanoconjugates containing both cetuximab and gemcitabine on a single gold nanoparticle core | Cetuximab helps in the targeted delivery of gemcitabine to the EGFR positive cancer | Ex vivo: pancreatic cancer cell lines (AsPC-1, PANC-1, and MIA Paca-2)
In vivo: orthotropic model of pancreatic cancer using nude mice | 2008 | [108] | |
| Lapatinib | Paclitaxel | Liposome | Lapatinib inhibits angiogenesis. Paclitaxel arrests cells at G2/M phase | Ex vivo: 4T1 mouse mammary carcinoma cells | 2015 | [124] | |
| Lapatinib | Paclitaxel | Polylactide-co-poly-(ethylene glycol) filomicelles of 100 nm length and spherical micelles of 20 nm diameter | Lapatinib inhibits angiogenesis and p-GP protein. Paclitaxel arrests cells at G2/M phase | Ex vivo: MCF-7 breast cancer cell | 2019 | [125] | |
| Gefitinib | DOX | Gefitinib complexed with dioleoyl-phosphatidic acid via ion paring was loaded onto the nanoparticle made of DOX conjugated poly(L-lactide)-block-polyethylene glycol (PLA-b-PEG) | At first, Gefitinib was released, followed by DOX. Gefitinib inhibits EGFR tyrosine kinase and DOX kills cancer cells | Ex vivo: MDA-MB-468 (breast cancer cell line)
In vivo: orhrotropic breast cancer model using FVB female mice and R7 murine breast cancer cells | 2017 | [126] | |
| Gefitinib | Gemcitabine | Gemcitabine was administered intravenously in saline solution. Gefitinib was dissolved in water and administered as oral gavage | Gefitinib inhibits EGFR tyrosine kinases and gemcitabine kills cancer cells | Ex vivo: UMSCC-1 cell line
In vivo: nude mice bearing UMSCC-1 xenografts | 2006 | [127] | |
| Erlotinib and Fedratinib | Not applicable | Poly(ethylene glycol)-poly (lactic acid) nanoparticle | Inhibition of EGFR and suppression of the JAK2/STAT3 signalling pathway | Ex vivo: nonsmall cell lung cancer (H1650, H1975)
In vivo: subcutaneous tumour-bearing male athymic nude mice | 2018 | [128] | |
| Lapatinib | Paclitaxel | Polylactide-co-Poly(ethylene glycol) micelles | Lapatinib inhibits EGFR and HER2 tyrosine kinase whereas paclitaxel arrests cancer cells at G2/M phase | Ex vivo: MCF-7 breast cancer cell line | 2019 | [129] | |
| Lapatinib | Paclitaxel | Liposome | Lapatinib inhibits EGFR and HER2 tyrosine kinase whereas paclitaxel arrests cancer cells at G2/M phase | Ex vivo: 4T1 murine mammary cell | 2016 | [130] | |
| Afatinib | Paclitaxel | Afatinib was loaded in stearic acid-based solid lipid nanoparticles. This nanoparticle and paclitaxel were loaded in polylactide-coglycolide-based porous microspheres | Afatinib inhibits EGFR and HER2 tyrosine kinase whereas paclitaxel arrests cancer cells at G2/M phase | Ex vivo: drug-resistant NSCLC | 2019 | [131] | |
| Erlotinib | Paclitaxel | Both erlotinib and paclitaxel were encapsulated in glyceryl monostearate nanoparticles, which was coated with a PEGylated polymeric layer | Erlotinib inhibits EGFR tyrosine kinase whereas paclitaxel arrests cancer cells at G2/M phase | Ex vivo: NCI-H23 cell line | 2018 | [132] | |
| Erlotinib | Gemcitabine | Erlotinib (100 mg/d, orally), Gemcitabine (1000 mg/m2, i.v. infusion) | Erlotinib inhibits EGFR tyrosine kinase whereas gemcitabine kills cancer cells | Clinical (open level phase II clinical trial): patients with locally advanced, inoperable, or metastatic pancreatic cancer | 2013 | [133] | |
| Erlotinib | DOX | pH-sensitive charge conversion nanocarrier. DOX was loaded in amino-functionalized mesoporous silica nanoparticles, which was coated with a synthetic zwitterionic oligopeptide lipid-containing erlotinib | Erlotinib and DOX were released sequentially and showed a synergistic effect. Erlotinib inhibits EGFR tyrosine kinase whereas DOX kills cancer cells | Ex vivo: A549 cell line
In vivo: tumour xenograft model using SD rats | 2016 | [134] | |
| Androgen receptor | Thalidomide | Not applicable | Methoxy poly(ethylene glycol)-poly(ε-caprolactone) nanoparticle | Thalidomide inhibits androgen receptor and TNF-α | Ex vivo: A549 cell line
In vivo: A549 xenograft model in nude mice | 2018 | [135] |
| mTOR | Everolimus | Not applicable | Everolimus loaded 3’-(1-carboxy)ethyl sialyl LewisX mimic-decorated liposome | Sialyl LewisX (sLeX), the natural ligand of E-selectin directs the delivery of liposome to tumour endothelium. Everolimus inhibits angiogenesis | Ex vivo: human umbilical vein endothelial cells | 2019 | [136] |
| Everolimus | Paclitaxel | Poly(ethylene glycol)-b-poly(lactide-coglycolide) copolymer nanoparticle. Everolimus:Paclitaxel molar ratio = 0.5:1 | Everolimus suppresses tumour growth by antiangiogenic effect. Paclitaxel kills the cancer cells | Ex vivo: different breast cancer cell lines like MDA-MB-231, MDA-MB-468, MCF-7, TrR1, MDA-MB-231-H2N and SKBR3 | 2018 | [137] | |
| Rapamycin | Cisplatin | Nanoprecipitate of cisplatin was coated with di-oleoyl-phosphatidic acid. It was further encapsulated in PLGA nanoparticles. Rapamycin was dispersed in PLGA shell | Rapamycin inhibits tumour growth by the antiangiogenic effect. It promotes vascular normalization to improve tumour perfusion. Thus the tumour cells are sensitized to cytotoxic cisplatin molecule | Ex vivo: A375 melanoma cells
In vivo: xenograft model of human melanoma | 2014 | [138] |
List of FDA-approved anti-angiogenic agents for the treatment of cancer
| Serial No. | Agents | Marketed name | Mechanism | FDA approved therapy | Ref. |
|---|---|---|---|---|---|
| 1. | Afatinib | Gilotrif® | Inhibits EGFR (ErbB1), HER2 (ErbB2), and HER4 (ErbB4) receptors | 1st-line treatment of patients with metastatic NSCLC (Jan 12, 2018) | [139] |
| 2. | Axitinib and pembrolizumab | Inlyta® and Keytruda® | Axitinib inhibits tyrosine kinase 1, 2 and 3 of VEGFR. Pembrolizumab binds to the Programmed cell death protein 1 (PD-1) receptor, blocking both immune-suppressing ligands, PD-L1 and PD-L2, from interacting with PD-1 to help restore T-cell response and immune response against cancer cells | Advanced renal cell carcinoma (Jan 27, 2017) | [140] |
| 3. | Bevacizumab | Avastin® | It acts by selectively binding circulating VEGF, thereby inhibiting the binding of VEGF to its cell surface receptors. This inhibition leads to a reduction in microvascular growth of tumour blood vessels and thus limits the blood supply to tumour tissues | Avastin was approved for the most aggressive form of brain cancer (Dec 5, 2017), metastatic cervical cancer (Aug 14, 2014), and breast cancer (Nov 18, 2011).
Avastin in combination with 5-FU was approved for metastatic carcinoma of the colon and rectum (Feb 26, 2004). Avastin plus chemotherapy has been approved for the initial treatment of metastatic non-squamous, NSCLC (Dec 6, 2018), women with advanced ovarian cancer following initial surgery (Jun 13, 2018), platinum-resistant recurrent ovarian cancer (Nov 14, 2014), first-line treatment of most common types of lung cancer (Oct 11, 2006). Avastin in combination with paclitaxel chemotherapy for first-line treatment of advanced HER2-negative breast cancer (Feb 25, 2008) | [141] |
| 4. | Bosutinib | Busulif® | It is an ATP-competitive Bcr-Abl tyrosine-kinase inhibitor with an additional inhibitory effect on SRC family kinases (including Src, Lyn and Hck). It is also active against the receptors for PDGF and VEGF | Philadelphia chromosome-positive (Ph+) CML with resistance, or intolerance to prior therapy (Sep 5, 2012) | [142] |
| 5. | Cabozantinib | Cabometyx® and Cometriq® | It is a multiple tyrosine kinase inhibitor
(c-Met, VEGFR2, AXL and RET receptor) | Advanced renal cell carcinoma (Feb 15, 2018), renal cell carcinoma and hepatocellular carcinoma (Apr 25, 2016) | [143,144] |
| 6. | Cetuximab | Erbitux® | Epidermal growth factor receptor inhibitor | Squamous cell carcinoma of the head and neck (Mar 2016) | [145] |
| 7. | Crizotinib | Xalkori® | Inhibitor of receptor tyrosine kinases including ALK, hepatocyte growth factor receptor (HGFR, c-Met), and RON | NSCLC (Aug 26, 2011) | [146] |
| 8 | Dasatinib | Sprycel® | It is a dual Bcr-Abl and Src family tyrosine kinase inhibitor. It also targets tyrosine kinases of EPHA2, PDGFR, GFR, and c-KIT | Paediatric patients with Philadelphia chromosome-positive (Ph+) CML in the chronic phase (Nov 9, 2017) | [147] |
| 9. | Erlotinib | Tercava® | It inhibits the intracellular phosphorylation of tyrosine kinase associated with the EGFR | Lung and pancreatic cancer (Nov 18, 2004) | [148] |
| 10. | Everolimus | Afinitor® | Inhibitor of mTOR | Renal cell carcinoma, breast cancer, neuroendocrine carcinoma (Mar 30, 2009) | [149] |
| 11. | Gefitinib | Iressa® | Selective inhibitor of the EGFR | NSCLC (May 2003) | [150] |
| 12. | Imatinib | Gleevec® | Protein-tyrosine kinase inhibitor that inhibits the Bcr-Abl tyrosine kinase, the constitutive abnormal tyrosine kinase created by the Philadelphia chromosome abnormality in CML | Acute lymphoblastic leukaemia, chronic myelogenous leukaemia, myelodysplastic diseases, gastrointestinal stromal tumour (May 10, 2001) | [151] |
| 13. | Lapatinib with Capecitabine | Tykerb® | Dual tyrosine kinase inhibitor which interrupts the HER2/neu and EGFR pathways | Breast cancer (Mar 13, 2007) | [152] |
| 14. | Lenalidomide | Revlimid® | Directly and indirectly by inhibition of bone marrow stromal cell support, by anti-angiogenic and anti-osteoclastogenic effects | Follicular lymphoma (May 28, 2019) | [153] |
| 15. | Nilotinib | Tasigna® | Acts as TKI and blocks a tyrosine kinase protein called Bcr-Abl | CML (Mar 22, 2018) | [154] |
| 16. | Nintedanib | Ofev® and Vargatef® | It binds to the intracellular ATP binding pockets of FGFR 1-3, PDGFRα/β, and VEGFR 1-3. This results in blockage of the autophosphorylation of these receptors and the downstream signalling cascades | Idiopathic pulmonary fibrosis (2014) | [155] |
| 17. | Osimertinib | Tagrisso® | It targets the mutated EGFR T790M within the cancer cells | NSCLC (Apr 2018) | [156] |
| 18. | Pazopanib | Votrient® | It inhibits VEGFR, PDGFR, c-KIT and FGFR | Advanced soft tissue sarcoma (Apr 27, 2012) | [157] |
| 19. | Ponatinib | Iclusig® | It inhibits Bcr-Abl, an abnormal tyrosine kinase that is the hallmark of CML and Ph+ ALL | Adult patients with chronic phase, accelerated phase, or blast phase CML or Ph+ ALL for whom no other TKI therapy is indicated (Dec 14, 2012) | [158] |
| 20. | Ramucirumab | Cyramza® | It is a direct VEGFR2 antagonist, that binds with high affinity to the extracellular domain of VEGFR2 and block the binding of natural VEGFR ligands (VEGF-A, VEGF-C and VEGF-D) | Gastric cancer, NSCLC, colorectal cancer, hepatocellular carcinoma (Apr 21, 2014) | [159] |
| 21. | Regorafenib | Stivarga® | Dual targeted VEGFR2 and Tie2 tyrosine kinase inhibition | Hepatocellular carcinoma (Apr 27, 2017)
Advanced gastrointestinal stromal tumour (Feb 25, 2013) Advanced colorectal cancer (Sep 27, 2012) | [160] |
| 22. | Sorafenib | Nexavar® | Protein kinase inhibitor with activity against many protein kinases, including VEGFR, PDGFR and RAF kinases | Advanced renal cell carcinoma (Dec 20, 2005) | [161] |
| 23. | Sunitinib | Sutent® | Multi-targeted RTK inhibitor | Renal cell carcinoma (Nov 16, 2017) | [162] |
| 24. | Temsirolimus | Torisel® | Inhibitor of mTOR | Renal cell carcinoma (May 30, 2007) | [163] |
| 25. | Thalidomide | Thalomid® | Inhibitor of Akt phosphorylation | Multiple myeloma (May 26, 2006) | [164] |
| 26. | Vandetanib | Caprelsa® | It inhibits EGFR | Advanced thyroid cancer (Apr, 2011) | [165] |
| 27. | Ziv- aflibercept | Zaltrap® | It is a recombinant protein that strongly binds with VEGFR and blocks all known ligands for this receptor | Colorectal cancer (Aug 15, 2012) | [166] |
Enhancement of vasculature permeability by physical treatment
EPR is a highly heterogeneous phenomenon. It is variable, even amongst different regions of the same tumour. In fact, within a single tumour, not all blood vessels are permeable to the same extent. Moreover, in many clinical settings, it has been found that tumours do not have a sufficient level of EPR to ensure the accumulation of nanomedicines. This is mainly because of the insufficient permeability of the vascular endothelium of tumour blood vessels. This problem can be addressed by local application of physical treatments such as sonoporation, hyperthermia, and radiotherapy that enhance tumour vasculature permeability, and aid in extravasation of nanomedicines uniformly throughout the TME.
Sonoporation
Sonoporation involves the application of ultrasonic sound to increase the gap between vascular endothelial cells. The mechanical effects can be further augmented with microbubbles and nanobubbles. The acoustic waves generate acoustic radiation force that causes bulk streaming and microstreaming. Bulk streaming is the movement of localized fluid current in the direction of propagation of ultrasonic sound while microstreaming involves localized eddies that are generated next to cavitating bodies. All these mechanical outputs may result in the release of drugs from carriers and the associated movement of drug molecules into targeted tissues. The efficiency of drug release is controlled by acoustic parameters like ultrasound frequency, power density, and pulse duration. Gas-filled micro-bubbles and nano-bubbles undergo violent collapse under large acoustic pressures. This phenomenon is known as inertial cavitation and is responsible for the generation of micro-streaming[167,168], shock waves[169-174], and jetting which are all responsible for enhancing the effect of EPR. The stability of bubbles is mainly affected by the transport properties of core gas. Air, and biologically inert heavy gases like sulphur hexafluoride, perfluorocarbon are used mainly. Though microbubbles are more responsive to ultrasonic radiation and undergo large changes in volume for the induction of EPR, they cannot escape the capillaries. In contrast, nanobubbles can easily penetrate the tumour via EPR. High-frequency ultrasound is thus suitable for targeted delivery of therapeutic agents to small and superficial tumours, whereas low-frequency ultrasound is beneficial for the treatment of large and deeply located ones.
Theek et al.[175] studied the effect of sonoporation and softshell/hardshell microbubbles on tumour accumulation of fluorophore-labelled 100 nm liposomes in mice bearing A431, BxPC-3 tumour. There was a 100% enhancement in tumour accumulation of liposome.
In another study, Yan et al.[176] attached paclitaxel encapsulated liposomes to the lipid shell of microbubbles via avidin-biotin linkage. They achieved high encapsulation efficiency of doxorubicin and upon application of ultrasonic sound of optimized intensity for the optimal period of time, there was significant enhancement in the uptake of drug molecules in 4T1 breast tumours by EPR.
As an alternative approach, Meng et al.[177] developed a doxorubicin loaded nanobubble [Figure 5]. It consisted of a core of a polymeric network where doxorubicin is dispersed. This core was encapsulated in a perfluoropropane gas bubble, the lipid shell of which was further stabilized with pluronic molecules. When delivered intravenously in combination with therapeutic ultrasonication, this ~170 nm diameter nanobubble showed higher accumulation and better distribution of doxorubicin in tumours, leading to significantly higher intracellular uptake and therapeutic efficacy.
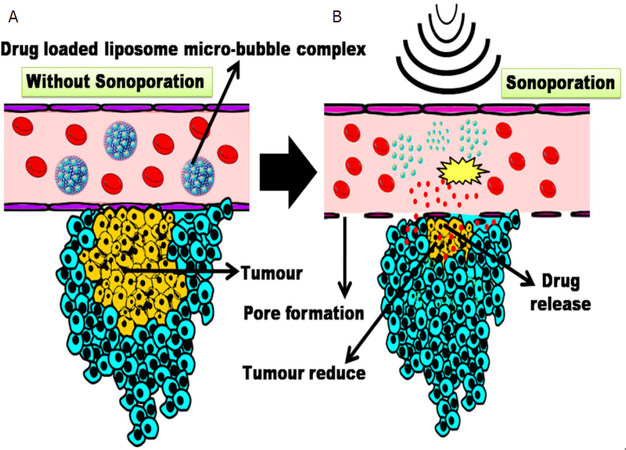
Figure 5. Schematic representation of cancer treatment with anticancer drug-loaded liposome-micro-bubble complexes (PLMC) assisted by ultrasound (US). A: when flowing through the target region, drugs remain attached to the lipid shells of MBs but are unable to cross the tumour vasculature by simple diffusion; B: application of high-intensity focused US bursts the micro-bubbles to release drugs. The cavitating and imploding MBs also enhance permeability of the plasma membrane, leading to higher uptake of released drugs. MBs: micro-bubbles
Hyperthermia
In response to temperatures of 41-45 °C, there is increased tissue perfusion to dissipate heat. For healthy tissues like muscle and skin, this increase in perfusion can be as high as 10- and 15-fold respectively.
In tumour tissue, perfusion rates are increased by 1.5-2 folds only[178,179]. Due to this insufficient perfusion, the temperature of tumour tissues raises further. This causes shut down of local blood flow due to (1) endothelial denaturation; (2) vasoconstriction in large pre-existing arterioles at the tumour periphery; and (3) increase in flow resistance because of high viscosity due to the formation of thrombus and fibrinogen gel. Ultimately, tumour cells are killed due to heat only.
Controlled, local heating of tumour tissue with radiofrequency[180], microwave or ultrasound to temperatures between 40-45 °C has the following effects: (1) dilatation of tumour vessels leading to enhanced blood flow; (2) enhancement in microvascular permeability to macromolecules[181] and nanomedicine[181,182]. This further increases the EPR effect; and (3) triggering the release of cargo molecules (therapeutic agents) from thermoresponsive nanomedicine[179].
There are different well-studied thermoresponsive nanomedicines such as liposomes[183-188], nanogels[189-192], hydrogel coated metal nanoparticles[193], polymeric nanoparticles[194-197] and elastin-like peptide-drug conjugates[179]. Thermodox® is a doxorubicin loaded thermoresponsive liposome, approved for the treatment of liver cancer. It is capable of delivering 25 times more doxorubicin to tumour tissues compared to intravenous infusion, and 5 times more doxorubicin than standard/ordinary liposomal formulation[23].
Again, to control drug release at mild hyperthermia, leucine zipper peptide was incorporated into the liposome[24]. At ~42 °C, the leucine zipper gate dissociated to release the drug precisely.
The thermo-responsive bubble generating liposomes[24] was also developed [Figure 6]. It consists of an ammonium bicarbonate loaded core, which generates CO2 upon application of hyperthermia (42 °C) and increases the permeability of the liposome bilayer by triggering the release of the drug.
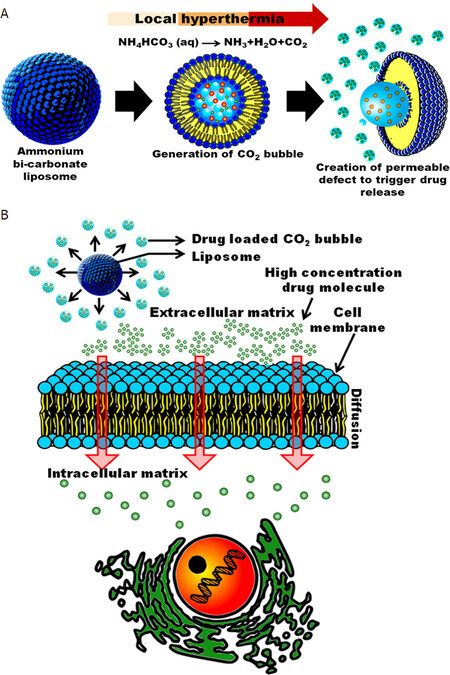
Figure 6. Schematic diagram showing the structure and function of thermoresponsive, bubble-generating liposomes and the mechanism of localized extracellular drug release triggered by heat. A: drug release mechanism upon application of hyperthermia; B: internalization of the released drug by the target cell
Gold nanoparticles coated with thermo-responsive hydrogel was developed for cancer therapy[198,199]. Local hyperthermia enhances the accumulation of nanoparticles within the tumour[200]. The gold nanoparticle has strong plasmon absorption, resulting in the generation of heat and removal of the polymeric shell. Thus, the gold nanoparticle acts as an anticancer agent[201,202].
Sato et al.[203] successfully applied threefold strategies to chemotherapy with Fe (Salen) nanoparticle. After intravenous injection, this magnetic nanoparticle was guided to the tumour site for delivery in a rabbit toung tumour model. The nanoparticle, at the target site, was heated with an alternating magnetic field for the local induction of hyperthermia that helped in further distribution of the nanoparticle into the TME due to the EPR effect.
Hyperthermia by NIR laser irradiation causes shrinkage of blood vessels and tumour ablation. Combining hyperthermia and chemotherapy could be an efficient treatment approach. This is known as photothermal chemotherapy[204]. Docetaxel loaded polypyrrole and hyaluronic acid-modified phospholipid nanoparticle were used for photothermal chemotherapy[205]. There was complete inhibition of tumours in 4T1 tumour-bearing mice.
Whole-body hyperthermia at the mild fever range (39.5 °C, for 4-6 h) was found to help in the therapeutic efficacy of doxorubicin-loaded liposome in syngeneic CT26 colorectal mice carcinoma[206]. There was a threefold increase in drug uptake in the tumour. It was also reported to be associated with decreased IFP and an increased fraction of perfused microvessels[207].
Concluding remarks
Hypoxia-induced formation of new blood vessels is the key factor in the progression of tumours. Tumour vasculature is heterogeneous, tortuous, irregularly branched, and hyperpermeable. Due to poor lymphatic drainage, the TME has high IFP. This heterogeneity of the vasculature, high IFP, poor extravasation due to sluggish blood flow, and larger distance between exchange vessels are all potential barriers to the delivery of therapeutic agents to tumours. A rationally designed delivery system should overcome all these barriers to reach deep tumour tissue. As the endothelial cells of tumour vasculature have longer gaps, and the IFP is high, nanoparticles of proper size can inherently be accumulated in the tumour due to the EPR effect. This is known as passive targeting. The surface of nanocarriers can also be coated with monoclonal antibodies against receptor proteins overexpressed in proangiogenic tumour cells for active targeted drug delivery. The vascular barrier can be further reduced by enhancing blood perfusion in the tumour and normalization of tumour vasculature. Local delivery of mediators such as NO and CO enhance blood perfusion whereas inhibition of proangiogenic pathways and the use of antiangiogenic agents help in the accumulation of anticancer drugs loaded nanocarriers deep within tumour tissues. Furthermore, the use of sonoporation and hyperthermia boosts nanocarrier mediated tumour-targeted drug delivery.
Declarations
AcknowledgmentsThe authors are grateful to the Guru Nanak Institute of Pharmaceutical Science & Technology and Department of Biotechnology, University of Calcutta for providing literature resources and other software facilities required for writing the manuscript.
Authors’ contributionsContributed in writing the manuscript: Dastidar DG
Contributed in editing the manuscript: Chakrabarti G
Did the literature survey and prepared the diagrams: Ghosh D
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
2. Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis: good and evil. Genes Cancer 2011;2:1117-33.
3. Nagy JA, Dvorak HF. Heterogeneity of the tumor vasculature: the need for new tumor blood vessel type-specific targets. Clin Exp Metastasis 2012;29:657-62.
4. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 2011;10:417-27.
5. Nagy JA, Feng D, Vasile E, Wong WH, Shih SC, et al. Permeability properties of tumor surrogate blood vessels induced by VEGF-A. Lab Invest 2006;86:767-80.
6. Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol 2000;156:1363-80.
7. McDonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res 2002;62:5381-5.
8. Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev 2007;26:281-90.
10. Liu QL, Liang QL, Li ZY, Zhou Y, Ou WT, et al. Function and expression of prolyl hydroxylase 3 in cancers. Arch Med Sci 2013;9:589-93.
11. Koh MY, Darnay BG, Powis G. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1alpha, leading to its oxygen-independent degradation. Mol Cell Biol 2008;28:7081-95.
12. Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J Am Chem Soc 2012;134:4465-8.
13. Abboud MI, McAllister TE, Leung IKH, Chowdhury R, Jorgensen C, et al. 2-Oxoglutarate regulates binding of hydroxylated hypoxia-inducible factor to prolyl hydroxylase domain 2. Chem Commun (Camb) 2018;54:3130-3.
14. Mandl M, Depping R. Hypoxia-inducible aryl hydrocarbon receptor nuclear translocator (ARNT) (HIF-1β): is it a rare exception? Mol Med 2014;20:215-20.
15. Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, et al. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci U S A 1996;93:12969-73.
16. Van Hove CE, Van der Donckt C, Herman AG, Bult H, Fransen P. Vasodilator efficacy of nitric oxide depends on mechanisms of intracellular calcium mobilization in mouse aortic smooth muscle cells. Br J Pharmacol 2009;158:920-30.
17. Pereira KM, Chaves FN, Viana TS, Carvalho FS, Costa FW, et al. Oxygen metabolism in oral cancer: HIF and GLUTs (Review). Oncol Lett 2013;6:311-6.
18. Chaneton B, Gottlieb E. PGAMgnam style: a glycolytic switch controls biosynthesis. Cancer Cell 2012;22:565-6.
19. Ye F, Chen Y, Xia L, Lian J, Yang S. Aldolase A overexpression is associated with poor prognosis and promotes tumor progression by the epithelial-mesenchymal transition in colon cancer. Biochem Biophys Res Commun 2018;497:639-45.
20. Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med 2017;9:eaao4583.
21. Ahmad SS, Glatzle J, Bajaeifer K, Bühler S, Lehmann T, et al. Phosphoglycerate kinase 1 as a promoter of metastasis in colon cancer. Int J Oncol 2013;43:586-90.
22. Skuli N, Liu L, Runge A, Wang T, Yuan L, et al. Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2alpha) alters vascular function and tumor angiogenesis. Blood 2009;114:469-77.
23. Takagi H, Koyama S, Seike H, Oh H, Otani A, et al. Potential role of the angiopoietin/tie2 system in ischemia-induced retinal neovascularization. Invest Ophthalmol Vis Sci 2003;44:393-402.
24. Zhang L, Yang N, Park JW, Katsaros D, Fracchioli S, et al. Tumor-derived vascular endothelial growth factor up-regulates angiopoietin-2 in host endothelium and destabilizes host vasculature, supporting angiogenesis in ovarian cancer. Cancer Res 2003;63:3403-12.
25. Gu J, Yamamoto H, Ogawa M, Ngan CY, Danno K, et al. Hypoxia-induced up-regulation of angiopoietin-2 in colorectal cancer. Oncol Rep 2006;15:779-83.
27. Sevick EM, Jain RK. Measurement of capillary filtration coefficient in a solid tumour. Cancer Res 1991;51:1352-5.
28. Gamble J, Smaje LH, Spencer PD. Filtration coefficient and osmotic reflection coefficient to albumin in rabbit submandibular gland capillaries. J Physiol 1988;398:15-32.
29. Gerlowski LE, Jain RK. Effect of hyperthermia on microvascular permeability to macromolecules in normal and tumor tissues. Int J Microcirc Clin Exp 1985;4:363-72.
31. Dvorak HF, Nagy JA, Dvorak JT, Dvorak AM. Identification and characterization of the blood vessels of solid tumors that are leaky to circulating macromolecules. Am J Pathol 1988;133:95-109.
32. Jain RK. Transport of molecules in the tumour interstitium: a review. Cancer Res 1987;47:3039-51.
33. Boucher Y, Baxter LT, Jain RK. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Res 1990;50:4478-84.
34. Wiig H, Tveit E, Hultborn R, Reed RK, Weiss L. Interstitial fluid pressure in DMBA-induced rat mammary tumours. Scand J Clin Lab Invest 1982;42:159-64.
35. Gerlowski LE, Jain RK. Microvascular permeability of normal and neoplastic tissues. Microvasc Res 1986;31:288-305.
36. Khawar IA, Kim JH, Kuh HJ. Improving drug delivery to solid tumors: Priming the tumour microenvironment. J Controll Release 2015;201:78-89.
37. Butler TP, Grantham FH, Gullino PM. Bulk transfer of fluid in the interstitial compartment of mammary tumors. Cancer Res 1975;35:3084-8.
38. Braun RD, Abbas A, Bukhari SO, Wilson W. Hemodynamic parameters in blood vessels in choroidal melanoma xenografts and rat choroid. Invest Ophthalmol Vis Sci 2002;43:3045-52.
39. Chary SR, Jain RK. Direct measurement of interstitial convection and diffusion of albumin in normal and neoplastic tissues by fluorescence photobleaching. Proc Natl Acad Sci U S A 1989;86:5385-9.
40. Wiig H, Tenstad O, Iversen PO, Kalluri R, Bjerkvig R. Interstitial fluid: the overlooked component of the tumor microenvironment? Fibrogenesis Tissue Repair 2010;3:12.
41. Wagner M, Wiig H. Tumour interstitial fluid formation, characterization, and clinical implications. Front Oncol 2015;5:115.
42. Jain RK, Baxter LT. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: significance of elevated interstitial pressure. Cancer Res 1988;48:7022-32.
43. Clauss MA, Jain RK. Interstitial transport of rabbit and sheep antibodies in normal and neoplastic tissues. Cancer Res 1990;50:3487-92.
44. Golombek SK, May JN, Theek B, Appold L, Drude N, et al. Tumor targeting via EPR: strategies to enhance patient responses. Adv Drug Deliv Rev 2018;130:17-38.
45. Salvioni L, Rizzuto MA, Bertolini JA, Pandolfi L, Colombo M, et al. Thirty years of cancer nanomedicine: success, frustration, and hope. Cancers (Basel) 2019;11:1855.
46. Zanotelli MR, Reinhart-King CA. Mechanical forces in tumor angiogenesis. Adv Exp Med Biol 2018;1092:91-112.
47. Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci U S A 2012;109:15101-8.
48. Zuo H. iRGD: a promising peptide for cancer imaging and a potential therapeutic agent for various cancers. J Oncol 2019;2019:9367845.
49. Deshpande PP, Biswas S, Torchilin VP. Current trends in the use of liposomes for tumour targeting. Nanomedicine (Lond) 2013;8:1509-28.
51. Chen Z, Zheng Y, Shi Y, Cui Z. Overcoming tumor cell chemoresistance using nanoparticles: lysosomes are beneficial for (stearoyl) gemcitabine-incorporated solid lipid nanoparticles. Int J Nanomedicine 2018;13:319-36.
52. Duan X, He C, Kron SJ, Lin W. Nanoparticle formulations of cisplatin for cancer therapy. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2016;8:776-91.
53. Krens SD, Lassche G, Jansman FGA, Desar IME, Lankheet NAG, et al. Dose recommendations for anticancer drugs in patients with renal or hepatic impairment. Lancet Oncol 2019;20:e200-7.
54. De Angelis C. Side effects related to systemic cancer treatment: are we changing the Promethean experience with molecularly targeted therapies? Curr Oncol 2008;15:198-9.
55. Golombek SK, May JN, Theek B, Appold L, Drude N, et al. Tumor targeting via EPR: strategies to enhance patient responses. Adv Drug Deliv Rev 2018;130:17-38.
56. Danhier F, Lecouturier N, Vroman B, Jérôme C, Marchand-Brynaert J, et al. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Control Release 2009;133:11-7.
57. Lu Z, Yeh TK, Tsai M, Au JL, Wientjes MG. Paclitaxel-loaded gelatin nanoparticles for intravesical bladder cancer therapy. Clin Cancer Res 2004;10:7677-84.
58. Zamboni WC. Liposomal, nanoparticle, and conjugated formulations of anticancer agents. Clin Cancer Res 2005;11:8230-4.
59. Hu H, Wang B, Lai C, Xu X, Zhen Z, et al. iRGD-paclitaxel conjugate nanoparticles for targeted paclitaxel delivery. Drug Dev Res 2019;80:1080-8.
60. Mangaiyarkarasi R, Chinnathambi S, Karthikeyan S, Aruna P, Ganesan S. Paclitaxel conjugated Fe3O4@LaF3:Ce3+,Tb3+ nanoparticles as bifunctional targeting carriers for cancer theranostics application. J Magnetism Magnetic Materials 2016;399:207-15.
61. Dalela M, Shrivastav TG, Kharbanda S, Singh H. pH-sensitive biocompatible nanoparticles of paclitaxel-conjugated poly(styrene-co-maleic acid) for anticancer drug delivery in solid tumors of syngeneic mice. ACS Appl Mater Interfaces 2015;7:26530-48.
62. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986;46:6387-92.
63. Laitakari J, Näyhä V, Stenbäck F. Size, shape, structure, and direction of angiogenesis in laryngeal tumour development. J Clin Pathol 2004;57:394-401.
64. Hillen F, Griffioen AW. Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev 2007;26:489-502.
65. Ziyad S, Iruela-Arispe ML. Molecular mechanisms of tumor angiogenesis. Genes Cancer 2011;2:1085-96.
66. Azzopardi EA, Ferguson EL, Thomas DW. The enhanced permeability retention effect: a new paradigm for drug targeting in infection. J Antimicrob Chemother 2013;68:257-74.
67. Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer 2004;4:806-13.
68. Holdman XB, Welte T, Rajapakshe K, Pond A, Coarfa C, et al. Upregulation of EGFR signaling is correlated with tumor stroma remodeling and tumor recurrence in FGFR1-driven breast cancer. Breast Cancer Res 2015;17:141.
69. Dastidar DG, Das A, Datta S, Ghosh S, Pal M, et al. Paclitaxel-encapsulated core-shell nanoparticle of cetyl alcohol for active targeted delivery through oral route. Nanomedicine (Lond) 2019;14:2121-50.
70. Sun Q, Ojha T, Kiessling F, Lammers T, Shi Y. Enhancing tumor penetration of nanomedicines. Biomacromolecules 2017;18:1449-59.
71. Zhang YR, Lin R, Li HJ, He WL, Du JZ, et al. Strategies to improve tumor penetration of nanomedicines through nanoparticle design. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2019;11:e1519.
72. Nagamitsu A, Greish K, Maeda H. Elevating blood pressure as a strategy to increase tumor-targeted delivery of macromolecular drug SMANCS: cases of advanced solid tumors. Jpn J Clin Oncol 2009;39:756-66.
73. Leffler CW, Parfenova H, Jaggar JH. Carbon monoxide as an endogenous vascular modulator. Am J Physiol Heart Circ Physiol 2011;301:H1-11.
74. Suzuki M, Hori K, Abe I, Saito S, Sato H. A new approach to cancer chemotherapy: selective enhancement of tumor blood flow with angiotensin II. J Natl Cancer Inst 1981;67:663-9.
75. Scicinski J, Oronsky B, Ning S, Knox S, Peehl D, et al. NO to cancer: The complex and multifaceted role of nitric oxide and the epigenetic nitric oxide donor, RRx-001. Redox Biol 2015;6:1-8.
76. Frérart F, Sonveaux P, Rath G, Smoos A, Meqor A, et al. The acidic tumor microenvironment promotes the reconversion of nitrite into nitric oxide: towards a new and safe radiosensitizing strategy. Clin Cancer Res 2008;14:2768-74.
77. Tahara Y, Yoshikawa T, Sato H, Mori Y, Zahangir MH, et al. Encapsulation of a nitric oxide donor into a liposome to boost the enhanced permeation and retention (EPR) effect. Medchemcomm 2016;8:415-21.
78. Wei G, Wang Y, Huang X, Yang G, Zhao J, et al. Enhancing the accumulation of polymer micelles by selectively dilating tumor blood vessels with no for highly effective cancer treatment. Adv Healthc Mater 2018;7:e1801094.
79. Fang J, Islam R, Islam W, Yin H, Subr V, et al. Augmentation of EPR effect and efficacy of anticancer nanomedicine by carbon monoxide generating agents. Pharmaceutics 2019;11:343.
80. Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov 2010;9:728-43.
81. Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev 2008;60:79-127.
82. Fang J, Akaike T, Maeda H. Antiapoptotic role of heme oxygenase (HO) and the potential of HO as a target in anticancer treatment. Apoptosis 2004;9:27-35.
83. Fang J, Qin H, Nakamura H, Tsukigawa K, Shin T, et al. Carbon monoxide, generated by heme oxygenase-1, mediates the enhanced permeability and retention effect in solid tumors. Cancer Sci 2012;103:535-41.
84. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005;307:58-62.
86. Goedegebuure RSA, de Klerk LK, Bass AJ, Derks S, Thijssen VLJL. Combining radiotherapy with anti-angiogenic therapy and immunotherapy; a therapeutic triad for cancer? Front Immunol 2019;9:3107.
87. Sersa G, Jarm T, Kotnik T, Coer A, Podkrajsek M, et al. Vascular disrupting action of electroporation and electrochemotherapy with bleomycin in murine sarcoma. Br J Cancer 2008;98:388-98.
88. Teicher BA, Dupuis NP, Emi Y, Ikebe M, Kakeji Y, et al. Increased efficacy of chemo- and radio-therapy by a hemoglobin solution in the 9L gliosarcoma. In Vivo 1995;9:11-8.
89. Czito BG, Bendell JC, Willett CG, Morse MA, Blobe GC, et al. Bevacizumab, oxaliplatin, and capecitabine with radiation therapy in rectal cancer: Phase I trial results. Int J Radiat Oncol Biol Phys 2007;68:472-8.
90. Fitzgerald KA, O’Neill LAJ, Gearing AJH, Callard RE. The cytokine factsbook and webfacts In: Fitzgerald KA, Callard RE, editors. 2th ed. London: Academic Press; 2001. pp. 139-41.
91. O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 1997;88:277-85.
92. Bonneterre J, Montpas N, Boularan C, Galés C, Heveker N. Chapter Seven - analysis of arrestin recruitment to chemokine receptors by bioluminescence resonance energy transfer. In: Handel TM, editor. Methods in enzymology. Academic Press; 2016. pp. 131-53.
93. Kamphaus GD, Colorado PC, Panka DJ, Hopfer H, Ramchandran R, et al. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J Biol Chem 2000;275:1209-15.
94. Sund M, Nyberg P, Eikesdal HP. Endogenous matrix-derived inhibitors of angiogenesis. Pharmaceuticals 2010;3:3021-39.
95. Keith B, Simon MC. 17 - Tumour angiogenesis. In: Mendelsohn J, editor. The molecular basis of cancer 4th edition. Philadelphia: 2015. pp. 257-68.e2.
96. Singhal S, Mehta J. Thalidomide in cancer: potential uses and limitations. Bio Drugs 2001;15:163-72.
98. Méndez-Vidal MJ, Molina Á, Anido U, Chirivella I, Etxaniz O, et al. Pazopanib: evidence review and clinical practice in the management of advanced renal cell carcinoma. BMC Pharmacol Toxicol 2018;19:77.
99. Yoshida-Ichikawa Y, Tanabe M, Tokuda E, Shimizu H, Horimoto Y, et al. Overcoming the adverse effects of everolimus to achieve maximum efficacy in the treatment of inoperable breast cancer: a review of 11 cases at our hospital. Case Rep Oncol 2018;11:511-20.
100. Cai X, Zhu H, Zhang Y, Gu Z. Highly efficient and safe delivery of VEGF siRNA by bioreducible fluorinated peptide dendrimers for cancer therapy. ACS Appl Mater Interfaces 2017;9:9402-15.
101. YSaw PE, Zhang A, Nie Y, Zhang L, Xu Y, Xu X. Tumor-associated fibronectin targeted liposomal nanoplatform for cyclophilin A siRNA delivery and targeted malignant glioblastoma therapy. Front Pharmacol 2018;9:1194.
102. Xu X, Li Z, Zhao X, Keen L, Kong X. Calcium phosphate nanoparticles-based systems for siRNA delivery. Regen Biomater 2016;3:187-95.
103. Zheng G, Zhao R, Xu A, Shen Z, Chen X, et al. Co-delivery of sorafenib and siVEGF based on mesoporous silica nanoparticles for ASGPR mediated targeted HCC therapy. Eur J Pharm Sci 2018;111:492-502.
104. Shen J, Sun H, Meng Q, Yin Q, Zhang Z, et al. Simultaneous inhibition of tumor growth and angiogenesis for resistant hepatocellular carcinoma by co-delivery of sorafenib and survivin small hairpin RNA. Mol Pharm 2014;11:3342-51.
105. Li F, Wang Y, Chen WL, Wang DD, Zhou YJ, et al. Co-delivery of VEGF siRNA and etoposide for enhanced anti-angiogenesis and anti-proliferation effect via multi-functional nanoparticles for orthotopic non-small cell lung cancer treatment. Theranostics 2019;9:5886-98.
106. Fountzilas G, Kourea HP, Bobos M, Televantou D, Kotoula V, et al. Paclitaxel and bevacizumab as first line combined treatment in patients with metastatic breast cancer: the Hellenic Cooperative Oncology Group experience with biological marker evaluation. Anticancer Res 2011;31:3007-18.
107. Bartczak D, Muskens OL, Sanchez-Elsner T, Kanaras AG, Millar TM. Manipulation of in vitro angiogenesis using peptide-coated gold nanoparticles. ACS Nano 2013;7:5628-36.
108. Patra CR, Bhattacharya R, Wang E, Katarya A, Lau JS, et al. Targeted delivery of gemcitabine to pancreatic adenocarcinoma using cetuximab as a targeting agent. Cancer Res 2008;68:1970-8.
109. Lin YW, Raj EN, Liao WS, Lin J, Liu KK, et al. Co-delivery of paclitaxel and cetuximab by nanodiamond enhances mitotic catastrophe and tumor inhibition. Sci Rep 2017;7:9814.
110. Li F, Danquah M, Singh S, Wu H, Mahato RI. Paclitaxel- and lapatinib-loaded lipopolymer micelles overcome multidrug resistance in prostate cancer. Drug Deliv Transl Res 2011;1:420-8.
111. Yang Z, Xiang B, Dong D, Wang Z, Li J, et al. Dual receptor-specific peptides modified liposomes as VEGF siRNA vector for tumor-targeting therapy. Curr Gene Ther 2014;14:289-99.
112. Shein SA, Kuznetsov II, Abakumova TO, Chelushkin PS, Melnikov PA, et al. VEGF- and VEGFR2-targeted liposomes for cisplatin delivery to glioma cells. Mol Pharm 2016;13:3712-23.
113. Yao Y, Wang T, Liu Y, Zhang N. Co-delivery of sorafenib and VEGF-siRNA via pH-sensitive liposomes for the synergistic treatment of hepatocellular carcinoma. Artif Cells Nanomed Biotechnol 2019;47:1374-83.
114. Chen WH, Yang Sung S, Fadeev M, Cecconello A, Nechushtai R, et al. Targeted VEGF-triggered release of an anti-cancer drug from aptamer-functionalized metal-organic framework nanoparticles. Nanoscale 2018;10:4650-7.
115. Chen J, Sun X, Shao R, Xu Y, Gao J, et al. VEGF siRNA delivered by polycation liposome-encapsulated calcium phosphate nanoparticles for tumor angiogenesis inhibition in breast cancer. Int J Nanomedicine 2017;12:6075-88.
116. Doddapaneni R, Patel K, Owaid IH, Singh M. Tumor neovasculature-targeted cationic PEGylated liposomes of gambogic acid for the treatment of triple-negative breast cancer. Drug Deliv 2016;23:1232-41.
117. Yang ZZ, Li JQ, Wang ZZ, Dong DW, Qi XR. Tumor-targeting dual peptides-modified cationic liposomes for delivery of siRNA and docetaxel to gliomas. Biomaterials 2014;35:5226-39.
118. Yanagisawa M, Yorozu K, Kurasawa M, Nakano K, Furugaki K, et al. Bevacizumab improves the delivery and efficacy of paclitaxel. Anticancer Drugs 2010;21:687-94.
119. Dragovich T, Laheru D, Dayyani F, Bolejack V, Smith L, et al. Phase II trial of vatalanib in patients with advanced or metastatic pancreatic adenocarcinoma after first-line gemcitabine therapy (PCRT O4-001). Cancer Chemother Pharmacol 2014;74:379-87.
120. Lei M, Ma G, Sha S, Wang X, Feng H, et al. Dual-functionalized liposome by co-delivery of paclitaxel with sorafenib for synergistic antitumor efficacy and reversion of multidrug resistance. Drug Deliv 2019;26:262-72.
121. Yang X, Li H, Qian C, Guo Y, Li C, et al. Near-infrared light-activated IR780-loaded liposomes for anti-tumor angiogenesis and Photothermal therapy. Nanomedicine 2018;14:2283-94.
122. He J, Xiao H, Li B, Peng Y, Li X, et al. The programmed site-specific delivery of the angiostatin sunitinib and chemotherapeutic paclitaxel for highly efficient tumor treatment. J Mater Chem B 2019;7:4953-62.
123. Szlachcic A, Pala K, Zakrzewska M, Jakimowicz P, Wiedlocha A, et al. FGF1-gold nanoparticle conjugates targeting FGFR efficiently decrease cell viability upon NIR irradiation. Int J Nanomedicine 2012;7:5915-27.
124. Dehghan Kelishady P, Saadat E, Ravar F, Akbari H, Dorkoosh F. Pluronic F127 polymeric micelles for co-delivery of paclitaxel and lapatinib against metastatic breast cancer: preparation, optimization and in vitro evaluation. Pharm Dev Technol 2015;20:1009-1017.
125. Zajdel A, Wilczok A, Jelonek K, Musiał-Kulik M, Foryś A, et al. Cytotoxic effect of paclitaxel and lapatinib co-delivered in polylactide-co-poly(ethylene glycol) micelles on HER-2-negative breast cancer cells. Pharmaceutics 2019;1:169.
126. Zhou Z, Jafari M, Sriram V, Kim J, Lee JY, et al. Delayed sequential co-delivery of gefitinib and doxorubicin for targeted combination chemotherapy. Mol Pharm 2017;14:4551-9.
127. Chun PY, Feng FY, Scheurer AM, Davis MA, Lawrence TS, et al. Synergistic effects of gemcitabine and gefitinib in the treatment of head and neck carcinoma. Cancer Res 2006;66:981-8.
128. Chen D, Zhang F, Wang J, He H, Duan S, et al. Biodegradable nanoparticles mediated co-delivery of erlotinib (ELTN) and fedratinib (FDTN) toward the treatment of ELTN-resistant non-small cell lung cancer (NSCLC) via suppression of the JAK2/STAT3 signaling pathway. Front Pharmacol 2018;9:1214.
129. Zajdel A, Wilczok A, Jelonek K, Musiał-Kulik M, Foryś A, et al. Cytotoxic effect of paclitaxel and lapatinib co-delivered in polylactide-co-poly(ethylene glycol) micelles on HER-2-negative breast cancer cells. Pharmaceutics 2019;11:169.
130. Ravar F, Saadat E, Kelishadi PD, Dorkoosh FA. Liposomal formulation for co-delivery of paclitaxel and lapatinib, preparation, characterization and optimization. J Liposome Res 2016;26:175-87.
131. Yang Y, Huang Z, Li J, Mo Z, Huang Y, et al. PLGA porous microspheres dry powders for codelivery of afatinib-loaded solid lipid nanoparticles and paclitaxel: novel therapy for EGFR tyrosine kinase inhibitors resistant nonsmall cell lung cancer. Adv Healthc Mater 2019;8:e1900965.
132. Gupta B, Poudel BK, Regmi S, Pathak S, Ruttala HB, et al. Paclitaxel and erlotinib-co-loaded solid lipid core nanocapsules: assessment of physicochemical characteristics and cytotoxicity in non-small cell lung cancer. Pharm Res 2018;35:96.
133. Vaccaro V, Bria E, Sperduti I, Gelibter A, Moscetti L, et al. First-line erlotinib and fixed dose-rate gemcitabine for advanced pancreatic cancer. World J Gastroenterol 2013;19:4511-9.
134. He Y, Su Z, Xue L, Xu H, Zhang C. Co-delivery of erlotinib and doxorubicin by pH-sensitive charge conversion nanocarrier for synergistic therapy. J Control Release 2016;229:80-92.
135. Chen LX, Ni XL, Zhang H, Wu M, Liu J, et al. Preparation, characterization, in vitro and in vivo anti-tumor effect of thalidomide nanoparticles on lung cancer. Int J Nanomedicine 2018;13:2463-76.
136. Chantarasrivong C, Higuchi Y, Tsuda M, Yamane Y, Hashida M, et al. Sialyl LewisX mimic-decorated liposomes for anti-angiogenic everolimus delivery to E-selectin expressing endothelial cells. RSC Advances 2019;9:20518-27.
137. Houdaihed L, Evans JC, Allen C. Codelivery of paclitaxel and everolimus at the optimal synergistic ratio: a promising solution for the treatment of breast cancer. Mol Pharm 2018;15:3672-81.
138. Guo S, Lin CM, Xu Z, Miao L, Wang Y, et al. Co-delivery of cisplatin and rapamycin for enhanced anticancer therapy through synergistic effects and microenvironment modulation. ACS Nano 2014;8:4996-5009.
139. US-FDA. FDA broadens afatinib indication to previously untreated, metastatic NSCLC with other non-resistant EGFR mutations. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-broadens-afatinib-indication-previously-untreated-metastatic-nsclc-other-non-resistant-egfr [Last accessed on 12 Apr 2020].
140. US-FDA. FDA approves pembrolizumab plus axitinib for advanced renal cell carcinoma. Available from: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-plus-axitinib-advanced-renal-cell-carcinoma [Last accessed on 12 Apr 2020].
141. US-FDA. Avastin Approval History. Available from: https://www.drugs.com/history/avastin.html [Last accessed on 12 Apr 2020].
142. US-FDA. FDA grants accelerated approval to bosutinib for treatment of newly-diagnosed PH+ CML. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-bosutinib-treatment-newly-diagnosed-ph-cml [Last accessed on 12 Apr 2020].
143. US-FDA. FDA Approved Uses of Cabozantinib. Available from: https://www.cancernetwork.com/thyroid-cancer/fda-approved-uses-cabozantinib [Last accessed on 12 Apr 2020].
144. US-FDA. FDA approves cabozantinib for hepatocellular carcinoma. Available from: https://www.fda.gov/drugs/fda-approves-cabozantinib-hepatocellular-carcinoma [Last accessed on 12 Apr 2020].
145. US-FDA. Information on Cetuximab (marketed as Erbitux). Available from: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/information-cetuximab-marketed-erbitux [Last accessed on 12 Apr 2020].
146. US-FDA. FDA Approves Crizotinib Capsules. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-crizotinib-capsules [Last accessed on 12 Apr 2020].
147. US-FDA. FDA approves dasatinib for pediatric patients with CML. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-dasatinib-pediatric-patients-cml [Last accessed on 12 Apr 2020].
148. US-FDA. FDA approves Erlotinib (Tarceva) as first-line lung cancer therapy for certain patients. Available from: https://www.cancernetwork.com/lung-cancer/fda-approves-erlotinib-tarceva-first-line-lung-cancer-therapy-certain-patients [Last accessed on 12 Apr 2020].
149. US-FDA. Everolimus (Afinitor). Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/everolimus-afinitor [Last accessed on 12 Apr 2020].
150. Zeneca A. IRESSA® (gefitinib) approved by the U.S. Food and Drug Administration for first-line treatment of advanced EGFR mutation-positive non-small cell lung cance. Available from: https://https://www.astrazeneca.com/media-centre/press-releases/2015/iressa-fda-approved-non-small-cell-lung-cancer-treatment-13072015.html# [Last accessed on 12 Apr 2020].
151. US-FDA. FDA gives fast approval to gleevec in treatment of CML. Available from: https://www.cancernetwork.com/chronic-myeloid-leukemia/fda-gives-fast-approval-gleevec-treatment-cml [Last accessed on 12 Apr 2020].
152. Ryan Q, Ibrahim A, Cohen MH, Johnson J, Ko CW, et al. FDA drug approval summary: lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 2008;13:1114-9.
153. US-FDA. FDA approves lenalidomide for follicular and marginal zone lymphoma. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-lenalidomide-follicular-and-marginal-zone-lymphoma [Last accessed on 12 Apr 2020].
154. US-FDA. FDA approves nilotinib for pediatric patients with newly diagnosed or resistant/intolerant Ph+ CML in chronic phase. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-nilotinib-pediatric-patients-newly-diagnosed-or-resistantintolerant-ph-cml-chronic [Last accessed on 12 Apr 2020].
155. US-FDA. FDA approves first treatment for patients with rare type of lung disease. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-patients-rare-type-lung-disease [Last accessed on 12 Apr 2020].
156. US-FDA. FDA approves osimertinib for first-line treatment of metastatic NSCLC with most common EGFR mutations. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-osimertinib-first-line-treatment-metastatic-nsclc-most-common-egfr-mutations [Last accessed on 12 Apr 2020].
157. US-FDA. FDA approves Pazopanib for advanced soft-tissue sarcoma. Available from: https://www.ascopost.com/issues/may-15-2012/fda-approves-pazopanib-for-advanced-soft-tissue-sarcoma/ [Last accessed on 12 Apr 2020].
158. US-FDA. Ponatinib (marketed as Iclusig) Informaton. Available from: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/ponatinib-marketed-iclusig-informaton [Last accessed on 12 Apr 2020].
159. US-FDA. FDA approves ramucirumab for hepatocellular carcinoma. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ramucirumab-hepatocellular-carcinoma [Last accessed on 12 Apr 2020].
160. US-FDA. Regorafenib becomes first FDA-approved drug for liver cancer in nearly a decade. Available from: https://www.cancer.gov/news-events/cancer-currents-blog/2017/fda-regorafenib-liver [Last accessed on 12 Apr 2020].
161. Kane RC, Farrell AT, Saber H, Tang S, Williams G, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res 2006;12:7271-8.
162. US-FDA. FDA approves sunitinib malate for adjuvant treatment of renal cell carcinoma. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-sunitinib-malate-adjuvant-treatment-renal-cell-carcinoma [Last accessed on 12 Apr 2020].
163. Kwitkowski VE, Prowell TM, Ibrahim A, Farrell AT, Justice R, et al. FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 2010;15:428-35.
164. US-FDA. Thalidomide OK’d for multiple myeloma: FDA approves thalidomide with strict rules to prevent birth defects. Available from: https://www.webmd.com/cancer/multiple-myeloma/news/20060526/thalidomide-okd-for-multiple-myeloma [Last accessed on 12 Apr 2020].
165. US-FDA. Caprelsa Approval History. Available from: https://www.drugs.com/history/caprelsa.html [Last accessed on 12 Apr 2020].
166. US-FDA. FDA Approves Zaltrap. Available from: https://www.drugs.com/newdrugs/fda-approves-zaltrap-metastatic-colorectal-cancer-3413.html [Last accessed on 12 Apr 2020].
167. Peruzzi G, Sinibaldi G, Silvani G, Ruocco G, Casciola CM. Perspectives on cavitation enhanced endothelial layer permeability. Colloids Surf B Biointerfaces 2018;168:83-93.
168. Sutton JT, Haworth KJ, Pyne-Geithman G, Holland CK. Ultrasound-mediated drug delivery for cardiovascular disease. Expert Opin Drug Deliv 2013;10:573-92.
169. Wu M, Wang Y, Wang Y, Zhang M, Luo Y, et al. Paclitaxel-loaded and A10-3.2 aptamer-targeted poly(lactide-co-glycolic acid) nanobubbles for ultrasound imaging and therapy of prostate cancer. Int J Nanomedicine 2017;12:5313-30.
170. Fan CH, Wang TW, Hsieh YK, Wang CF, Gao Z, et al. Enhancing boron uptake in brain glioma by a boron-polymer/microbubble complex with focused ultrasound. ACS Appl Mater Interfaces 2019;11:11144-56.
171. Cao Y, Chen Y, Yu T, Guo Y, Liu F, et al. Drug release from phase-changeable nanodroplets triggered by low-intensity focused ultrasound. Theranostics 2018;8:1327-39.
172. Zhang C, Huang P, Zhang Y, Chen J, Shentu W, et al. Anti-tumor efficacy of ultrasonic cavitation is potentiated by concurrent delivery of anti-angiogenic drug in colon cancer. Cancer Lett 2014;347:105-13.
173. Zhao YZ, Lin Q, Wong HL, Shen XT, Yang W, et al. Glioma-targeted therapy using Cilengitide nanoparticles combined with UTMD enhanced delivery. J Control Release 2016;224:112-25.
174. Park J, Aryal M, Vykhodtseva N, Zhang YZ, McDannold N. Evaluation of permeability, doxorubicin delivery, and drug retention in a rat brain tumor model after ultrasound-induced blood-tumor barrier disruption. J Control Release 2017;250:77-85.
175. Theek B, Baues M, Ojha T, Möckel D, Veettil SK, et al. Sonoporation enhances liposome accumulation and penetration in tumors with low EPR. J Control Release 2016;231:77-85.
176. Yan F, Li L, Deng Z, Jin Q, Chen J, et al. Paclitaxel-liposome-microbubble complexes as ultrasound-triggered therapeutic drug delivery carriers. J Control Release 2013;166:246-55.
177. Meng M, Gao J, Wu C, Zhou X, Zang X, et al. Doxorubicin nanobubble for combining ultrasonography and targeted chemotherapy of rabbit with VX2 liver tumor. Tumour Biol 2016;37:8673-80.
179. Ghosh Dastidar D, Chakrabarti G. Chapter 6 - Thermoresponsive Drug Delivery Systems, Characterization and Application. In: Mohapatra SS, Ranjan S, Dasgupta N, Mishra RK, Thomas S, editors. Applications of Targeted Nano Drugs and Delivery Systems. Amsterdam: Elsevier; 2019. pp. 133-55.
180. Elming PB, Sørensen BS, Oei AL, Franken NAP, Crezee J, et al. Hyperthermia: the optimal treatment to overcome radiation resistant hypoxia. Cancers (Basel) 2019;11:60.
181. Kong G, Braun RD, Dewhirst MW. Hyperthermia enables tumor-specific nanoparticle delivery: effect of particle size. Cancer Res 2000;60:4440-5.
182. Noguchi A, Takahashi T, Yamaguchi T, Kitamura K, Noguchi A, et al. Enhanced tumor localization of monoclonal antibody by treatment with kininase II inhibitor and angiotensin II. Jpn J Cancer Res 1992;83:240-3.
183. Liu P, Guo B, Wang S, Ding J, Zhou W. A thermo-responsive and self-healing liposome-in-hydrogel system as an antitubercular drug carrier for localized bone tuberculosis therapy. Int J Pharm 2019;558:101-9.
184. Dai M, Wu C, Fang HM, Li L, Yan JB, et al. Thermo-responsive magnetic liposomes for hyperthermia-triggered local drug delivery. J Microencapsul 2017;34:408-15.
185. Maekawa-Matsuura M, Fujieda K, Maekawa Y, Nishimura T, Nagase K, et al. LAT1-targeting thermoresponsive liposomes for effective cellular uptake by cancer cells. ACS Omega 2019;4:6443-51.
186. Zhou Q, You C, Ling Y, Wu H, Sun B. pH and thermo dual stimulus-responsive liposome nanoparticles for targeted delivery of platinum-acridine hybrid agent. Life Sci 2019;217:41-8.
187. Dai M, Wu C, Fang HM, Li L, Yan JB, et al. Thermo-responsive magnetic liposomes for hyperthermia-triggered local drug delivery. J Microencapsul 2017;34:408-15.
188. Shi D, Mi G, Shen Y, Webster TJ. Glioma-targeted dual functionalized thermosensitive Ferri-liposomes for drug delivery through an in vitro blood-brain barrier. Nanoscale 2019;11:15057-71.
189. Chang R, Tsai WB. Fabrication of photothermo-responsive drug-loaded nanogel for synergetic cancer therapy. Polymers (Basel) 2018;10:1098.
190. Singh A, Vaishagya K, K Verma R, Shukla R. Temperature/pH-triggered PNIPAM-based smart nanogel system loaded with anastrozole delivery for application in cancer chemotherapy. AAPS Pharm Sci Tech 2019;20:213.
191. Chen J, Wu M, Veroniaina H, Mukhopadhyay S, Li J, et al. Poly (N-isopropylacrylamide) derived nanogels demonstrated thermosensitive self-assembly and GSH-triggered drug release for efficient tumor therapy. Polymer Chem 2019;10:4031-41.
192. Sreerenganathan M, Mony U, Rangasamy J. Thermo-responsive fibrinogen nanogels: a viable thermo-responsive drug delivery agent for breast cancer therapy? Nanomedicine (Lond) 2014;9:2721-3.
193. Kim JH, Lee T. Thermo-responsive hydrogel-coated nanoshells for in vivo drug delivery. J Biomed Pharmaceutical Engineering 2008;21:29-35.
194. Wang C, Li Y, Ma Y, Gao Y, Dong D, et al. Thermo responsive polymeric nanoparticles based on poly(2-oxazoline)s and tannic acid. Journal of Polymer Science Part A:. Polymer Chemistry 2018;56:1520-7.
195. Cammas S, Suzuki K, Sone C, Sakurai Y, Kataoka K, et al. Thermo-responsive polymer nanoparticles with a core-shell micelle structure as site-specific drug carriers. J Control Release 1997;48:157-64.
196. Rejinold NS, Muthunarayanan M, Divyarani VV, Sreerekha PR, Chennazhi KP, et al. Curcumin-loaded biocompatible thermoresponsive polymeric nanoparticles for cancer drug delivery. J Colloid Interface Sci 2011;360:39-51.
197. Seo HI, Cho AN, Jang J, Kim DW, Cho SW, et al. Thermo-responsive polymeric nanoparticles for enhancing neuronal differentiation of human induced pluripotent stem cells. Nanomedicine 2015;11:1861-9.
198. Arafa MG, El-Kased RF, Elmazar MM. Thermoresponsive gels containing gold nanoparticles as smart antibacterial and wound healing agents. Sci Rep 2018;8:13674.
199. Kim JH, Lee TR. Thermo- and pH-responsive hydrogel-coated gold nanoparticles. Chem Materials 2004;16:3647-51.
200. Vines JB, Yoon JH, Ryu NE, Lim DJ, Park H. Gold nanoparticles for photothermal cancer therapy. Front Chem 2019;7:167.
201. Farooq MU, Novosad V, Rozhkova EA, Wali H, Ali A, et al. Gold nanoparticles-enabled efficient dual delivery of anticancer therapeutics to hela cells. Sci Rep 2018;8:2907.
202. Chandran PR, Thomas RT. Chapter 14 - Gold Nanoparticles. In: Thomas S, Grohens Y, Ninan N, editors. Cancer drug delivery, in nanotechnology applications for tissue engineering. Oxford: William Andrew Publishing; 2015. pp. 221-37.
203. Sato I, Umemura M, Mitsudo K, Fukumura H, Kim JH, et al. Simultaneous hyperthermia-chemotherapy with controlled drug delivery using single-drug nanoparticles. Sci Rep 2016;6:24629.
204. Kim S, Moon MJ, Poilil Surendran S, Jeong YY. Biomedical applications of hyaluronic acid-based nanomaterials in hyperthermic cancer therapy. Pharmaceutics 2019;11:306.
205. Zhao T, Qin S, Peng L, Li P, Feng T, et al. Novel hyaluronic acid-modified temperature-sensitive nanoparticles for synergistic chemo-photothermal therapy. Carbohydr Polym 2019;214:221-33.
206. Winslow TB, Eranki A, Ullas S, Singh AK, Repasky EA, et al. A pilot study of the effects of mild systemic heating on human head and neck tumour xenografts: analysis of tumour perfusion, interstitial fluid pressure, hypoxia and efficacy of radiation therapy. Int J Hyperthermia 2015;31:693-701.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ghosh Dastidar D, Ghosh D, Chakrabarti G. Tumour vasculature targeted anti-cancer therapy. Vessel Plus 2020;4:14. http://dx.doi.org/10.20517/2574-1209.2019.36
AMA Style
Ghosh Dastidar D, Ghosh D, Chakrabarti G. Tumour vasculature targeted anti-cancer therapy. Vessel Plus. 2020; 4: 14. http://dx.doi.org/10.20517/2574-1209.2019.36
Chicago/Turabian Style
Ghosh Dastidar, Debabrata, Dipanjan Ghosh, Gopal Chakrabarti. 2020. "Tumour vasculature targeted anti-cancer therapy" Vessel Plus. 4: 14. http://dx.doi.org/10.20517/2574-1209.2019.36
ACS Style
Ghosh Dastidar, D.; Ghosh D.; Chakrabarti G. Tumour vasculature targeted anti-cancer therapy. Vessel Plus. 2020, 4, 14. http://dx.doi.org/10.20517/2574-1209.2019.36
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 29 clicks
Cite This Article 29 clicks

 Like This Article 10
likes
Like This Article 10
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.