
Atherosclerosis microbiome: upcoming target for vaccine and drug development

Abstract
Cardiovascular disease (CVD) is the leading cause of morbidity and mortality in adults and is one critical area of the medical sciences. Atherosclerosis is the main underlying pathology and is characterized by chronic inflammation of the arterial walls. The current treatment modalities for CVD target hypertension, hyperlipidemia and hemostasis, and suppress inflammation without directly addressing the origin of inflammation. Thus, many individuals with multiple classic risk factors for CVD do not experience acute ischemic events. Moreover, myocardial infarction and stroke continue to occur in up to two-thirds of all patients. Because many cardiovascular events have not been explained by genetics or other risk factors, and multiple epidemiologic studies have consistently suggested an infectious component, the introduction of entirely novel approaches for diagnostics and treatment that target infections are acutely needed. These complementary novel approaches addressing additional manageable risk factors such as infections will be based on the concept of personalized medicine to control CVD and achieve longevity, while also increasing the quality of life. There are a variety of avenues that could enable such novel approaches. These focus on the discovery and characterization of the infective component of atherosclerosis, the atherosclerosis microbiome. Specifically, we provide an update of the latest developments in the oral microbiome and its relation to CVD.
Keywords
Introduction
Cardiovascular disease (CVD) is the commonest cause of mortality and morbidity globally[1,2]. Compared with the past, global progress in extending life expectancy is forecasted to be slower from 2016 to 2040. This trend resulted from predicted slowed advances on key drivers including stagnated gains on cardiovascular diseases, which was a major factor in historical improvements in life expectancy[3].
Atherosclerosis is a chronic vascular inflammation associated with hypercholesterolemia, accumulation of lipids, hypertension, diabetes, smoking, smooth muscle cell proliferation, cell apoptosis, necrosis, fibrosis, and genetic factors. Atherosclerosis causes plaque accumulation, obstructing blood flow and contributes to acute ischemic events such as myocardial infarction or stroke. In these events, the arterial wall inflammatory lesion becomes destabilized, leading to plaque rupture and discharge of its necrotic core in the circulation, triggering coagulation and thrombosis. Such vulnerable plaques present the highest risk of acute events. The risk of atherosclerotic disease has been observed to be significantly higher in patients with periodontal disease, independently of other established risk factors.
Many CVD patients do not present any of the classical risk factors. Between 60%-70% of individuals with multiple cardiovascular disease risk factors have not experienced a cardiovascular event[4] and only 50% of the CVD patients have been shown to have elevated serum cholesterol[5]. While major statin trials report an average 28% reduction in low-density lipoprotein (LDL) cholesterol and a 31% reduction in relative risk, patients still have significant residual risk[6]. Likewise, myocardial infarction and stroke continue to occur in up to two-thirds of all patients, even after many of these factors are addressed[7]. This “forgotten” majority of patients leave wide open the door for exploration of risk factors that have not been adequately addressed to date.
While the importance of the traditional risk factors is well established, the data indicate additional factors contributing to atherogenesis. Infectious processes and products of the endogenous microbiome are capable to modulate atherosclerosis and its complications either directly, or indirectly, by eliciting local and systemic responses that potentiate atherogenesis. Here we will focus on bacterial infections as potential contributors to vascular inflammation, with an emphasis on periodontal pathogens as an established component of the atherosclerosis microbiome.
Current view of the infectious component of atherosclerosis. Association of periodontitis with CVD
The initiation of atherogenic process is typical for a chronic inflammatory disease. This process starts with recruitment of leukocytes from blood flow, mediated by a range of endothelial surface-expressed adhesins (more details in the study by Libby[8]). The endothelial activation and subsequent leukocyte recruitment/transmigration in tissue is in response to an activating stimulus, which includes microbial constituents.
Gingivitis and its advanced stage, chronic periodontitis, are the most prevalent microbial infections in man. Only in recent decades has the association between periodontal diseases and systemic conditions such as coronary heart disease and stroke became subject to investigation[9-16].
The largest genome network analysis (63,746 cases and 130,681 controls) identified lipid metabolism and inflammation as main pathways involved in the genetic predisposition to coronary artery disease (CAD). Specifically, the four most significant pathways mapping to putative genes involved in CAD are linked to lipid metabolism and inflammation, underscoring the causal role of these activities in the genetic etiology of CAD. However, the genetic variants strongly associated with CAD explain approximately only at most 10.6% of CAD heritability[17-19].
Infection is an understudied contributing factor in vascular inflammation. Nevertheless, infectious component as a risk factor is supported by an abundance of epidemiological evidence[20,21] and animal models. Multiple independent pathways of evidence already pinpoint inflammation as a key regulatory process that links multiple risk factors for atherosclerosis and its complications with altered arterial biology[22]. Vascular infection due to transient bacteremia, from leaky guts or periodontal lesions alike, can lead to persistent inflammation, including one due to an intracellular bacterial “privileged niche”. Indeed, blood levels of inflammation markers (e.g., C-reactive protein, CRP) have been associated with vascular risk factors and the prevalence and incidence of atherothrombotic CVD[23]. Consequently, hsCRP (a high sensitivity CRP test) is an important prognostic factor for atherosclerosis[24].
More critical evidence can be found in the Northern Manhattan study of stroke incidence and prognosis. This prospective cohort study (1625 participants, mean age 68.5 ± 10.1 years; 64.9% women) demonstrated that infectious burden is associated with established measure of risk of stroke, carotid plaque intima-media thickness[25-27]. Interestingly, a measure of infectious burden associated with risk of atherosclerosis and stroke was independently associated in this study with cognitive performance. This demonstrated that infections may be a culprit in cognitive impairment as well[28].
Similarly, the Oral Infections and Vascular Disease Epidemiology Study (INVEST) demonstrated a direct relationship between tooth loss and carotid plaque prevalence[29] Specifically, colonization with pathogenic periodontal pathogens was associated with carotid artery intima-media thickness (IMT), a measure of subclinical vascular disease[30]. INVEST also demonstrated that severe periodontal bone loss was associated with a nearly 4-fold increase in risk for the presence of carotid atheroma [odds ratio (OR) 3.64, P < 0.05][31]. The study also provided data supporting an effect of the subgingival periodontal bacteria level and both systolic and diastolic blood pressure in addition to prevalence of hypertension[32]. At a protein level, higher secretory phospholipase A2 activity (an inflammatory enzyme associated with atherosclerosis) at high tertile of etiologic presence presents a mechanistic explanation of the link between periodontal bacteria and CVD[33].
In the Atherosclerosis Risk in Communities (ARIC) study of 8,363 men and women from four United States communities (aged 52 to 75 years), patients with both high attachment loss and high tooth loss [OR = 1.5, 95% confidence interval (CI): 1.1 to 2.0] and also edentulous individuals (OR = 1.8, 95%CI: 1.4 to 2.4) had elevated odds of prevalent coronary heart disease (CHD) in comparison with controls with low attachment loss and low tooth loss. A number of traditional risk factors for CHD were factored in. The ARIC results thus presented evidence that both tooth loss and periodontal disease (PD) are associated with prevalent CHD[34].
The Periodontitis and Its Relation to Coronary Artery Disease compared 805 patients (< 75 years of age) with myocardial infarction (MI) and 805 age-, sex- (male 81%), and area-matched controls (mean age 62 ± 8) without MI. This study determined that periodontitis was more common (43%) in patients than in controls (33%; P < 0.001). A significant increased risk for MI was observed in periodontitis patients (OR adjusted for confounders, 1.28; 95%CI)[35].
In a Polish case-control study, the level of PD was significantly associated with the risk of acute MI (OR = 2.4, 95%CI: 1.1 to 5.2, P = 0.0203). This was even after an adjustment for age, sex, smoking, hypertension, diabetes, body-mass index, education and income[36]. Interestingly, severe infection was sometimes associated with MI only in females. Similarly in another study, severe periodontitis was more prevalent in female patients than female controls (14% vs. 4%, P = 0.005). An increased risk for severe periodontitis in female patients with a first MI was reported (adjusted OR = 3.72, 95%CI: 1.24 to 11.16, P = 0.005)[37].
Furthermore, multivariate analysis of coronary heart disease (CHD) individuals demonstrated a higher prevalence of oral diseases and lower compliance to oral disease prevention compared to healthy controls. The analysis showed a positive association between edentulousness (OR = 1.37, 95%CI: 1.02 to 1.85), the number of endodontic lesions (OR = 4.37, 95%CI: 1.69 to 11.28), chronic periodontitis (OR = 5.87, 95%CI: 1.17 to 29.4), and CHD[38].
Similarly, a 9-year follow-up study examined a possible correlation between the duration of periodontal disease state and cardiometabolic risk factors. The odds ratio for the presence of ≥ 1 cardiometabolic risk factor (hypertension, hyperglycemia, dyslipidemia or obesity) in individuals with a longitudinal presence of periodontal pockets for ≥ 6 years was significantly higher compared to individuals without periodontal pockets[39].
Finally, the recent Malmö Offspring Study is a population-based study using multivariable regression models to analyze the presence of carotid plaque and asymptomatic carotid plaque as related to measures of periodontal disease. This study demonstrated that the risk of developing a carotid plaque in study subjects with periodontitis was significantly higher compared to periodontitis-free subjects, with odds ratioof 1.75 (95%CI: 1.11 to 2.78)[40].
Not surprisingly, endodontic infections were also associated with vascular inflammations. For example, endodontic infection was associated with higher prevalence of CHD and initial endothelial damage[41,42]. In a cross-sectional study of the association between apical periodontitis (AP) and CVD using noninvasive methods, flow-mediated dilatation (FMD) was found to be significantly impaired in AP patients (mean = 4.9% ± 2.05%) in comparison with healthy individuals (mean = 9.74% ± 2.59%, P = 0.000). There was a statistically significant difference observed between carotid IMT of the AP group (mean = 0.64 ± 0.12 mm) and control group (mean = 0.54 ± 0.08 mm) (P = 0.000). Furthermore, there was a significant inverse correlation observed between c-IMT and FMD (rs = -0.381, P < 0.001). This indicated an impaired FMD and greater carotid IMT in AP patients, supporting an association between endodontic infection and cardiovascular inflammatio[43].
Seroepidemiology
Serological animal and cell culture studies provided evidence that bacterial infection, often by the red complex pathogen Porphyromonas gingivalis, a major etiologic agent of PD, emerges as a new, important factor for atherosclerosis[44]. Based on the accumulated epidemiological data, the infection hypothesis for initiation/exacerbation of atherosclerosis has already been established[45,46]. Specifically, periodontal inflammatory mediators were recognized as contributors to or triggers for systemic inflammatory responses. Subgingival periodontal infection demonstrated an increased risk of developing atherosclerosis in periodontal patients by 168%[47].
The ARIC study also presented an association between systemic antibody response to periodontal pathogens and coronary heart disease in ever and never smokers[48]. The latest ARIC data presented significant association between high gingival inflammation, tooth loss, severe tooth loss, and severe periodontitis with diabetes, coronary heart disease, hsCRP, and IL-6, while only severe disease was associated with stroke[49].
Furthermore, coronary disease was more common among seropositive for P. gingivalis subjects, relative to the seronegative (14.0% and 9.7%, respectively; P = 0.029). Hence, CHD was more prevalent in individuals with a high combined antibody response against Aggregatibacter actinomycetemcomitans and P. gingivalis than in those with a low response (17.4% and 11.1%, P = 0.026). When adjusted for age and several CHD risk factors, the subjects with a high combined antibody response had an OR of 1.5 (95%CI: 0.95 to 2.50, P = 0.077) for prevalent coronary disease. The combined antibody response was directly associated with prevalent CHD (P = 0.046) and inversely associated with the concentration of serum high-density lipoprotein (HDL) cholesterol (P = 0.050). This demonstrated that serum antibodies to major periodontal bacteria were associated with CHD[50].
In addition, the same group demonstrated that systemic exposure to P. gingivalis predicts incident stroke. Investigating seropositive subjects, it was found that they had a multivariate odds ratio of 1.6 (95%CI: 1.0 to 2.6) for stroke, compared with the seronegative subjects. Additionally, patients with a history of stroke or CHD at baseline contained more often P. gingivalis IgA than the controls, 79.7% vs. 70.2%. The seropositive subjects had an odds ratio of 2.6 (1.0 to 7.0) for secondary stroke, compared with the seronegative[51]. In the CVD-free individuals (n = 893), systemic exposure to P. gingivalis increased the risk of stroke as follows: compared to seronegative subjects, men and women that were IgG-seropositive for P. gingivalis presented a multivariate OR (95%CI) of 1.63 (1.06 to 2.50) and 2.30 (1.39 to 3.78) for stroke, respectively. Interestingly, higher OR was observed in males, who had never smoked. Compared to seronegative men, P. gingivalis IgA-seropositive men had a OR of 3.31 (1.31 to 8.40, P = 0.012) for stroke. There was no association found between antibody titers to A. actinomycetemcomitans and stroke, suggesting that the systemic exposure specifically to P. gingivalis may contribute to incident stroke[52].
These authors also presented data demonstrating that periodontitis also causes mild changes in HDL metabolism. These changes appear to be less severe than those occurring during the acute-phase response. Thus, periodontitis may reduce the anti-atherogenic properties of HDL, increasing the risk for CHD. Importantly, the HDL-mediated cholesterol efflux improved after periodontal treatment. More interestingly, this increase was significant (P < 0.05) among those patients whose CRP titers decreased (53.7% reduction, P = 0.015) and who were PCR-positive for A. actinomycetemcomitans[53].
In comparison, P. gingivalis induces HDL oxidation, impairing the atheroprotective function of HDL. P gingivalis likely makes it proatherogenic by raising a proinflammatory response via interaction with monocytes and macrophages[54]. Overall, the presence of A. actinomycetemcomitans and P. gingivalis, major causative organisms in periodontitis was shown to be the strongest determinant of the systemic antibody response to these pathogens[55].
Concerning serum antibodies, in coronary disease as well as in periodontal disease patients the antibody titers against P. gingivalis were the most prevalent. Hs-CRP test levels and antibody titers to P. gingivalis have been reported to be higher in periodontitis patients than in control subjects[56]. Interestingly, while periodontal patients were seropositive for both studied P. gingivalis strains, FDC381 and Su63, higher antibody titers to P. gingivalis Su63 only was observed in coronary disease patients. This finding indicates that specific genomic virulence determinants present in particular P. gingivalis strains may affect atherogenesis[57].
The association of P. gingivalis antibodies with mortality is however non-linear. In a specific study, mortality was highest for those just above the median anti-P. gingivalis response and a reduced risk was present among those with low or high titers of the antibody[58], suggesting that the efficiency of the immune response itself may be the key to control of the infection.
In a first 27-year long-term study of association of chronic oral infections in childhood with subclinical carotid atherosclerosis in adulthood in 755 participants, the infections were associated with adulthood IMT. The relative risk (RR) found was 1.95 (95%CI), especially elevated in boys, RR 2.25 (95%CI). The associations were independent of cardiovascular risk factors[59]. Specifically, the salivary IgA antibody levels to malondialdehyde acetaldehyde-modified low-density lipoprotein (MAA-LDL), Rgp44 (gingipain A hemagglutinin domain of P. gingivalis), and Aa-HSP60 (heat shock protein 60 of A. actinomycetemcomitans) were discovered to be elevated in stable-CAD and acute coronary syndrome patients when compared to CAD-healthy subjects[60]. Periodontal patients were characterized by higher levels of subgingival bacteria. The serum IgA/IgG burden indicated higher risk for acute coronary syndrome (OR = 1.84, 95%CI: 1.01 to 3.35 for IgA; OR = 1.87, 95%CI: 1.01 to 3.46 for IgG). This risk was independent of other cardiovascular risk factors (body mass index, number of teeth, subgingival bacterial levels and periodontal diagnosis)[61]. The serological differences in periodontitis patients may present risk factors for atherosclerosis. These seroepidemiological findings are consistent with an association between periodontitis and cardiovascular disease.
Association of bacteria with atheromatous tissue
Identification of periodontal pathogens in vascular tissue. While oral tissues are the primary sites for P. gingivalis infection, it has been long shown it can also enter the circulation daily through the microvasculature following tooth brushing and other dental procedures[62]. Routine procedures such as tooth extraction may also lead to transient bacteremia[63-65]. Periodontal biofilm bacteria are thus disseminated to large vessels. Consequently, bacterial DNA was detected in atheromas by PCR[66] where P. gingivalis was the most abundant pathogen compared to all others tested species[67]. Similarly, a high content of periodontal pathogens were detected in atheromatous arterial specimens from atherosclerosis patients. The pathogens were specifically detected within primary atheromatous lesions. Critically, most patients had severe periodontitis[68].
Using reverse transcription polymerase chain reaction, DNA from endodontic bacteria was identified in 20/36 (56%) of aortic aneurism tissue specimens and DNA from periodontal bacteria in 17/36 (47%) of these specimens[69]. Compared to cardiac bypass control samples, both ruptured and unruptured aneurysm specimens presented significantly more bacterial DNA (P = 0.003 and 0.001, respectively)[70]. Further, quantitation of DNA from periodontopathic bacteria using universal and species-specific TaqMan probe/primer sets demonstrated total bacterial DNA in 94.9%, and periodontopathic bacterial DNA in 92.3 % of the atherosclerotic plaques from periodontal disease patients[71]. Using sequence analysis of bacterial 16S rRNA libraries from atherosclerotic plaques, 23 bacterial species/phylotypes were identified, where 15 (60.9%) of the phylotypes were reported as yet uncultivable or as yet uncharacterized species[72]. P. gingivalis DNA was found in 21 of 91 (23%) samples taken from carotid endarterectomies[73].
More importantly, live invasive periodontal pathogens, P. gingivalis and A. actinomycetemcomitans were identified in a patient plaque[74]. In the same line of investigation, a large number of strains were cultivated from patient plaques, belonging to different species, mostly associated with periodontal biofilm, including P. gingivalis[75,76]. It will also be interesting to adapt to atheromas the recently communicated reverse-genomics-enabled cultivation and characterization of as-yet-uncultured species[77].
It has also been shown that more than 90% of all infections in the head and neck region can have an odontogenic origin[78]. Most recently, P. gingivalis proteinase gingipain was detected in 96% of the 53 brain tissue sections from Alzheimer’s patients[79], indicating overall systemic hematogenous spread of periodontal bacteria.
Effects of bacterial infection of vasculature
A variety of communications on animal experiments have suggested that bacterial infection may predispose to early atherosclerosis[80] and plaque instability[81]. In addition to passive dissemination by way of the bloodstream, bacteria may disseminate and cause low-grade focal infections due to their ability to invade and persist intracellularly. Low-grade infection presents mixed positive/negative results for infection, inflammation or pathogen identification since it requires prolonged culturing. Thus, using quantitative polymerase chain reaction, it was shown that bacterial DNA was present in atherosclerotic plaque and - of note - the amount of this DNA correlated with the amount of leukocytes in the atherosclerotic plaque[82].
Bacteria possess a profound ability to disrupt the host homeostasis. For example, infection with P. gingivalis induces procoagulant effects in human endothelial cells[83]. Very important, an invasive, but not a non-invasive, P. gingivalis strain accelerated atherosclerosis in a murine model[84], pointing to the significance of strain-specific genomic virulence determinants. Furthermore, it was found that P. gingivalis invasion (but not a non-invasive mutant) in ApoE (±) mice was critical for atherosclerosis progression[45].
Bacterial infection can also cause apoptosis in endothelia[85]. There is a large body of evidence that P. gingivalis has developed an elaborate proteolytic system composed of surface-located or secreted enzymes, Rgp and Kgp gingipains, which serve to provide these asaccharolytic bacteria with sole source of nutrients in the form of small peptides and amino acids, thus functioning as virulence factors leading to tissue destruction[86-88]. Consequently, the proteolytic activities of this infectious agent may also contribute to vascular disruption and subsequent obstruction of the lumen.
At the same time, P. gingivalis cytotoxic activities have been well characterized[89] and loss of cell adhesion properties with subsequent apoptotic cell death has been observed[90-92]. Even more aggravating, P. gingivalis efficiently activates coagulation factors, thus promoting platelet aggregation[93-96]. Thus, internalized destructive platelet-aggregating inflammatory agent such as P. gingivalis combined with macrophage infiltration in intimal regions would likely contribute to triggering apoptosis and formation of necrotic core, potentially leading to plaque weakening and rupture, folloed by triggering of the coagulation cascade, thrombosis and acute ischemic events.
The Rgp gingipains of P. gingivalis lyse lipoproteins producing 2 apoE fragments, as well as 2 apoB-100 fragments, in LDL, while the Kgp gingipain lyses HDL, induce reactive oxygen species (ROS) and degrade antioxidants. In addition, both Rgp and Kgp gingipains induce lipid peroxidation. Thus, P. gingivalis may affect the lipoproteins expression in blood, another facet of its contribution to atherogenesis[97]. Similarly, Pep19 from P. gingivalis HSP60 has a distinct ability to induce native-LDL oxidation which may serve as a plausible mechanism by which this peptide may drive epitope spreading to the neoantigen, i.e., oxidized LDL, in the pathogenesis of atherosclerosis[98].
During endodontic (apical PD) infection, ligation of toll-like receptors (TLRs) on phagocytes’ surface triggers activation of humoral and cellular responses and also phagocytosis, synthesis of ROS and production of inflammatory mediators, cytokines and matrix metalloproteinases. TLRs provide innate immune sensing of conserved pathogen-associated molecular patterns. TLR - mediated signaling also contribute importantly to cardiovascular disease. For a recent review of this particular subject, see[99].
Specifically, oxidative stress has been strongly involved in the pathogenesis of atherosclerosis[100]. The ROS-producing systems in the vasculature include reduced nicotinamide adenine dinucleotide phosphate oxidase, xanthine oxidase, the mitochondrial electron transport chain, and nitric oxide (NO) synthase. Oxidative stress due to ROS overproduction contributes to all stages of atherogenesis, from the plaque formation to the most critical stage, the plaque rupture[101-103]. All cardiovascular risk factors such as hypercholesterolemia, hypertension, diabetes mellitus, and smoking increase ROS and decrease endothelial NO synthesis[104].
Bacterial pathogens can cause oxidative stress via triggering LDL oxidation at the atherosclerotic lesion. In addition to hematogenous dissemination to the atheroma, bacteria may spread in the system while intracellular, since they survive and multiply in peripheral blood mononuclear cells[75]. They are also able to replicate and persist within vascular endothelial and smooth muscle cells (SMCs)[105]. Intracellular Chlamydophila pneumoniae infection has been shown to induce ROS in macrophages, endothelial and smooth muscle cells, causing oxidative stress[106,107]. This can lead to endothelial dysfunction, foam cell formation, SMC proliferation, platelet aggregation as well as cytokine, growth factor, and cell adhesion molecule production[101].
Periodontal bacteria are extensively studied in respect of oxidative stress. P. gingivalis-induced ROS production was shown to activate the NOD-like receptor family, thus increasing the aortic gene expression of Nod-like receptor family, pyrin domain containing 3 (NLRP3), pro-interleukin (IL)-1β, pro-IL-18 and pro-caspase-1[108].
Further, P. gingivalis increases the uptake of oxidized LDL, promoting the foam cell formation[109]. P. gingivalis also induces the synthesis and secretion into the vascular lumen of monocyte chemoattractant protein-1 (MCP-1), causing monocyte influx[110][Figure 1]. In addition, this organism induces apoptosis[91,92,111], including in the presence of ox-LDL[112]. The presence of an apoptotic core is a hallmark of the unstable plaque. It can eventually lead to plaque erosion, rupture and acute ischemic events. Finally, P. gingivalis may destabilize the plaque also by enhancing matrix metalloproteinase-9 activity and oxidative stress through impairing the selective autophagic clearance of damaged mitochondria[113].
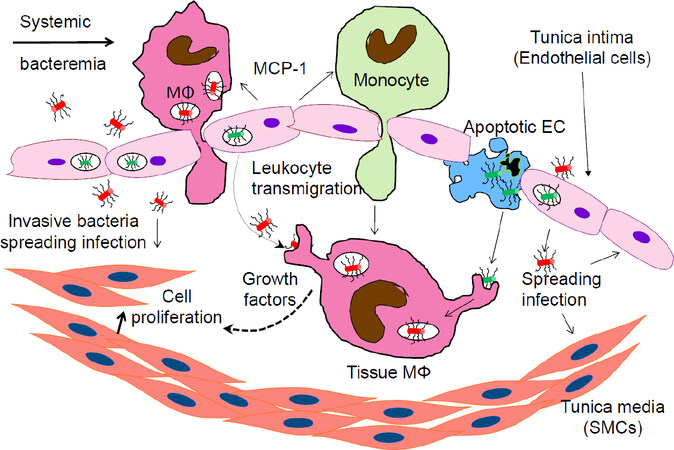
Figure 1. Bacterial component of atheromas representing transmigrating from the vascular lumen and tissue-embedded macrophages (MΦ). The activated inflammatory leukocytes roll over the intima, adhere to the endothelial cells and transmigrate in the tissue. This includes intracellular bacteria-carrying macrophages extravasating from the vascular lumen into the arterial wall. Bacteria induce host cells to release chemotaxis molecules (such as MCP-1) and growth factors stimulating host cell division. Invasive bacteria multiply, causing persistent inflammation and apoptotic cell death, forming a necrotic core and eroding the vascular wall. MN: extravasating monocyte; EC: endothelial cell; SMC: smooth muscle cell; MCP-1: monocyte chemoattractant protein-1
The LDL oxidation leads to an increased expression of adhesion molecules such as vascular cell adhesion molecule-1, intercellular adhesion molecule-1 and E-selectin on the activated endothelia. This results in the tethering of the leukocytes to the endothelia, adhesion and diapedesis (extravasation) into the vascular wall [Figure 1][114-116]. Thus, in a vicious circle, oxidative stress multiplies the effects of inflammation and accelerates the atherogenesis[117].
In another deleterious activity, the periodontal infection itself accelerates lipid deposition and atherosclerosis in animal models[118,119], and therefore addressing the infection would - as an important added benefit - suppress the atherogenic effects of dyslipidemia.
Atherogenic dyslipidemia (hypercholesterolaemia and hyperlipidaemia) is defined as high plasma LDL and low plasma HDL cholesterol with elevated triglycerides. The increase of triglyceride and LDL cholesterol levels include alterations observed in lipid metabolism and lipoprotein composition. Infection and the concomitant inflammation induce acute-phase response contributing to atherogenic changes in lipid and lipoprotein metabolism[120].
An early and consistent effect of infection/inflammation is increased serum triglyceride level, characterized by an increase in very low-density lipoprotein (VLDL) levels[121,122]. High bacterial lipopolysaccharide serum activity (endotoxemia) has shown a strong correlation with serum triglyceride concentrations (P < 0.001)[123]. The increase in serum triglycerides may be due to both an increase in hepatic VLDL production and a decrease in the clearance of triglyceride rich lipoproteins in chronic inflammation[124].
Endotoxemia also modulates HDL composition and size[122]. HDL is one of the plasma lipoproteins that neutralize Gram-negative bacterial LPS and Gram-positive bacterial lipoteichoic acid, thus favoring the clearance of these products[125]. For example, the mean percentage of HDL cholesterol in Helicobacter pylori-seropositive patients was significantly lower than the one measured in seronegative ones (P = 0.008 and P < 0.001, respectively)[126]. In another study, the Health 2000 Health Examination Survey, which included 8028 Finnish subjects aged 30 or older, no consistent association between serum lipid levels and periodontal infection among normoweight subjects was found. However, an association was found of high serum triglycerides and low HDL with periodontal infection among obese subjects[127]. Overall, these authors were not able to present evidence that unfavorable lipid composition can be considered as an important risk for periodontal infection in a general adult population[128].
Specifically, for periodontal infections, both total and LDL-cholesterol were significantly associated with antibody titer to P. gingivalis in non-obese patients[129]. The latest meta-analysis suggested that periodontitis is significantly associated with reduced HDL (P = 0.0005), elevated LDL (P = 0.003) and triglycerides (P < 0.0001) compared to healthy controls, supporting the rationale that periodontal disease is associated with lipid metabolic control[130].
Effect of periodontal treatment on atherosclerotic inflammation
Due to the extended amount of time during which the atherogenesis takes place and even more to obvious ethical reasons, it is virtually impossible to obtain an unambiguous demonstration of the effectivity of antibacterial treatment on atherosclerotic inflammation. This however should not prevent from exploring the alternative approaches to examine attenuation and moreover, reversal of atherogenesis via resolution of the vascular inflammation.
Current therapeutic strategies focus on anti-inflammation, i.e., on pharmacologic intervention in the inflammatory pathways. However, only resolution of inflammation will restore the homeostasis. The isolation and characterization of resolving agonist molecules using endogenous lipid mediators of resolution as potential therapeutic agents for the management of inflammation has opened a new promising area of research[131-134].
Thus, in an established rabbit model of aortic plaque development, it was shown that oral/topical application of a proresolution lipid mediator, Resolvin E1 (RvE1) diminished diet and periodontitis- induced aortic atherogenesis. Importantly, RvE1 not only significantly attenuated the arterial inflammation; the treatment also prevented periodontal inflammation (P < 0.05). In the absence of the latter, oral/topical administration of RvE1 led to a reduction of the arterial plaque and a lower intima-media thickness ratio. RvE1 also reduced the inflammatory cell infiltration in the animal model compared to non-treated controls (P < 0.001). In addition, local oral RvE1 application significantly diminished serum CRP levels (P < 0.05)[135,136].
Multiple clinical investigations have also indicated a positive effect of periodontal disease treatment on systemic inflammation. Periodontal therapy of periodontitis patients demonstrated significant improvements in periodontal pocket depth, in brachial artery FMD and in serum IL-6. A trend toward reduction in serum CRP has been reported in this study as well[137]. Similarly, after non-surgical treatment of periodontal patients, serum leptin, IL-6, and CRP levels were significantly decreased (mean ± SD before and after, P value, respectively: leptin, 8.02 ± 5.5, 7.10 ± 4.4, P = 0.015; IL-6, 1.73 ± 1.02, 1.36 ± 0.73, P = 0.048; and CRP, 802.0 ± 1065, 491.2 ± 479.3, P = 0.047)[138]. Further, in a study of 49 patients with moderate to advanced level of periodontitis, hsCRP and anti-P. gingivalis antibody levels were measured and the effect of periodontitis treatment, including surgery and use of antibacterials, was analyzed on both markers. The hsCRP levels and antibody titers were higher in the periodontitis patients than in the 40 periodontally healthy control subjects. Furthermore, periodontal treatment significantly decreased he CRP levels and the antibody titers (P < 0.005). A significant reduction of hs-CRP levels was communicated as a result of the treatment in patients with hs-CRP levels > 1 mg/L (P < 0.005)[56].
More data supporting treatment were provided by the INVEST study of 420 participants (68 ± 8 years old) at baseline and 3-year follow-up. The longitudinal carotid artery IMT progression was recorded. Adjustments were made for age, sex, race/ethnicity, diabetes, smoking status, education, body mass index, systolic blood pressure, LDL cholesterol and HDL cholesterol. An attenuation of the IMT progression was reported with improvement in clinical or microbial periodontal status[139].
A positive association has also been reported between severe periodontitis and oxidative stress[140]. Introduction of periodontal therapy triggered a burst of inflammatory response before a progressive and consistent reduction of systemic inflammation followed by an endothelial function improvement[141]. Furthermore, the reactive oxygen metabolites (d-ROMs) test values decreased and the biological antioxidant potential test values increased in patients with chronic apical periodontitis after endodontic therapy treatment. The oxidative stress levels in these patients exhibited a downtrend, returning to normal in 90 days post treatment[142]. Periodontal treatment however did not improve vasodilation in coronary disease patients in a brief follow-up period, even though it maintained the titers of vascular inflammation markers[143]. An in-depth review on oxidative stress in periodontal disease, focusing on the relationship between the local and systemic markers of oxidative stress and periodontal disease is in[144]. More viewpoints on the subject of treatment are reviewed in[145,146].
Finally, a meta-analysis was conducted investigating the literature on the association between carotid IMT (c-IMT), FMD and periodontitis. The effect of periodontal treatment on carotid IMT and FMD was assessed. Periodontal disease diagnosis was associated with a mean increase in c-IMT of 0.08 mm (95%CI: 0.07 to 0.09). The mean difference in FMD was 5.1% when compared to controls (95%CI: 2.08% to 8.11%). As a consequence of periodontal treatment, a mean improvement on FMD of 6.64% (95%CI: 2.83% to 10.44%) was observed between test and controls. This meta-analysis demonstrated an association between arterial inflammation, increased c-IMT and impaired FMD and PD. The results suggested a beneficial effect of periodontal treatment on FMD leading to improvement in endothelial function[147].
Exploring cutting-edge technologies to discover new therapeutic targets and approaches for drug development. Atherosclerosis microbiome
Presenting major fiscal burden, CVD is the costliest disease in the US at $555 billion (American Heart 2017, http://bit.ly/2LfsC5A) and €210 billion in the European Union (2017, http://bit.ly/2UDXS2F). This underscores the need for novel diagnostic and therapeutic developments since cost-effective and rapid approaches are lacking.
A major incentive for novel approaches is that many CVD events have not been explained by classical risk factors. To address a modifiable risk factor, such as microbial pathogens, Robert Koch’s postulate must be satisfied, namely the pathogen must be isolated from the diseased tissue. Thus, C. pneumoniae was isolated from a carotid endarterectomy specimen[148]. However, it took time before a polybacterial infectious component from atherosclerotic plaques was identified, and these clinical isolates cultivated. This achievement demonstrated the existence of atherosclerosis microbiome, a sample of the microbial community localized within human atheromatous tissues[67,74,75,149]. Such advancement enabled an entirely new approach to CVD diagnosis and treatment, fulfilling Koch’s postulate.
A natural approach to restore the homeostasis, reversing the atherogenic process, is via control of the inflammatory component, often originating from periodontal lesions. The latest network analysis confirmed inflammation and lipid metabolism as the two key biological pathways involved in the predisposition to CVD[150]. Of note, there is still no approach successfully addressing the actual source of the inflammation. Taking the potential opportunities presented by the identification of prokaryotes in vascular inflammations, randomized placebo-controlled clinical trials using antibiotics in the context of CVD have been designed[151,152]. However, the results were disappointing, since the administered treatment may not have reached its target (i.e., intracellular location sheltering bacteria from antibiotics as well as from immune response). The latest anti-inflammation trials (CIRT[153], CANTOS[154,155], TETHYS[156], SOLID-TIMI 52[157]) did not target the plausible origin of inflammation, intracellular bacteria. The PEGASUS-TIMI 54 trial, although also targeting ischemic events, was strictly thrombosis-related.
As mentioned before, both ethical considerations and the slow progress of atherosclerosis preclude conducting a clinical trial to establish causality. Importantly, the WIZARD trial design predicted that a positive effect of drugs would tighten the association between atherosclerosis and bacteria without proving causality[158]. As stated by Peter Libby, renowned vascular disease physician, “A therapeutic trial of antibiotics still would not establish a causal relationship between any particular infectious agent and atherosclerosis. Yet, if antibiotic treatment could reduce atherosclerosis events, the public health implications could be enormous; hence the need to keep an open mind”[159].
In addition, animal studies suggest that atherosclerosis is induced or aggravated by invasive infectious agents (i.e., persistent IC infection)[45,84,160-163].
It is becoming clear that atherosclerosis represents chronic vascular inflammation partly of microbial etiology. Intracellular invasive pathogens induce a low-grade persistent inflammation that exacerbate the atherogenesis. Therefore, to address intracellular infections as etiologic factors for CVD, entirely novel anti-infectives and vaccines are required[164]. Alleviating this key pathological feature (i.e., IC bacteria internalized in vascular tissue) could significantly improve the clinical outcome.
Evidence supporting this view is several-fold: (1) epidemiological data support an infectious component in atherosclerosis; (2) atherosclerosis has many of the characteristics of a chronic infectious disease; and (3) internalization of bacteria can produce a “privileged niche” (i.e., shelter from immune response and drugs)[165].
Of note, a variety of species, mostly with periodontal origin were cultivated from atheromatous tissue of endarterectomy patients[74-76]. These are promising targets for intervention, specifically P. gingivalis, a gram-negative anaerobe capable of invading a variety of non-phagocytic eukaryotic cells[166-170]. P. gingivalis is a key periodontal pathogen[171] causing inflammation and host tissue destruction. It becomes internalized and also persists in vascular cells[105]. While oral tissues are the primary sites for P. gingivalis infection, it can also enter the circulation daily through the microvasculature and its role in periodontitis is established[30]. Most importantly, an invasive P. gingivalis strain accelerated atherosclerosis in a murine model[84] and as mentioned before its tissue invasion ability was critical for atherosclerosis progression[45]. These advancements pave the way for further promising developments along the Alzheimer’s treatment technology where P. gingivalis is identified as a key target[79].
Addressing the atherosclerosis microbiome: a new approach to CVD risk modification
The cultivation and identification in atheromatous plaques of a variety of viable bacteria suggests that atherosclerotic lesions can be induced or exacerbated by these inflammatory pathogens. Importantly, it was shown that bacteria not only invaded both vascular cell types but also persisted intracellularly. Moreover, the bacteria were transmitted between both cell types and to healthy cells, explaining the chronicity of infection[105]. Such intracellular polymicrobial flora has been well demonstrated[172], pointing to a plausible contributor to premature atherosclerosis[173].
A natural approach to restore the homeostasis is reversing the atherogenic process, via control of the inflammatory component, often originating from periodontal lesions. An important advantage of such approach is that the main problem in medical care can be addressed as initiated or exacerbated by prokaryotes. Targeting bacteria, thus minimizing the side effects of treatment, is inherently more attractive than the current complicated designs addressing metabolic pathways.
Moreover, the infection itself accelerates lipid deposition and atherosclerosis in animal models[118,119], and therefore addressing the infection would also suppress the effects of hyperlipidemia.
A variety of available methodologies can be adapted to bring about development of vaccines and small molecule inhibitors of the identified pathogens. The emergence of infections, specifically from periodontal origin as a potential risk factor for CVD, is leading to a convergence in oral and medical care that will hopefully benefit the patients and public health[174].
Declarations
Authors’ contributionsWrote and reviewed the manuscript: Kozarov E, Progulske-Fox A
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Joseph P, Leong D, McKee M, Anand SS, Schwalm JD, et al. Reducing the global burden of cardiovascular disease, part 1: the epidemiology and risk factors. Circ Res 2017;121:677-94.
2. GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016;388:1459-544.
3. Foreman KJ, Marquez N, Dolgert A, Fukutaki K, Fullman N, et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet 2018;392:2052-90.
4. Greenland P, Knoll MD, Stamler J, Neaton JD, Dyer AR, et al. Major risk factors as antecedents of fatal and nonfatal coronary heart disease events. JAMA 2003;290:891-7.
6. Kones R. Molecular sources of residual cardiovascular risk, clinical signals, and innovative solutions: relationship with subclinical disease, undertreatment, and poor adherence: implications of new evidence upon optimizing cardiovascular patient outcomes. Vasc Health Risk Manag 2013;9:617-70.
7. Libby P. The forgotten majority: unfinished business in cardiovascular risk reduction. J Am Coll Cardiol 2005;46:1225-8.
8. Libby P. Inflammatory mechanisms: the molecular basis of inflammation and disease. Nutr Rev 2007;65:S140-6.
9. Seymour GJ, Ford PJ, Cullinan MP, Leishman S, Yamazaki K. Relationship between periodontal infections and systemic disease. Clin Microbiol Infect 2007;13:3-10.
10. Reyes L, Herrera D, Kozarov E, Roldá S, Progulske-Fox A. Periodontal bacterial invasion and infection: contribution to atherosclerotic pathology. J Periodontol 2013;84:S30-50.
11. Pietiäinen M, Liljestrand JM, Kopra E, Pussinen PJ. Mediators between oral dysbiosis and cardiovascular diseases. Eur J Oral Sci 2018;126:26-36.
12. Carrizales-Sepúlveda EF, Ordaz-Farías A, Vera-Pineda R, Flores-Ramírez R. Periodontal disease, systemic inflammation and the risk of cardiovascular disease. Heart Lung Circ 2018;27:1327-34.
13. Mesa F, Magan-Fernandez A, Castellino G, Chianetta R, Nibali L, et al. Periodontitis and mechanisms of cardiometabolic risk: Novel insights and future perspectives. Biochim Biophys Acta Mol Basis Dis 2019;1865:476-84.
14. Kozarov E, Progulske-Fox A. Pocket guide to bacterial infections. In: Balamurugan K, Udayakumar P, editors. Boca Raton FL: CRC Press; 2019. p. 398.
15. Bui FQ, Almeida-da-Silva CLC, Huynh B, Trinh A, Liu J, et al. Association between periodontal pathogens and systemic disease. Biomed J 2019;42:27-35.
16. Liccardo D, Cannavo A, Spagnuolo G, Ferrara N, Cittadini A, et al. Periodontal disease: a risk factor for diabetes and cardiovascular disease. Int J Mol Sci 2019;20:1414.
17. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, et al; CARDIoGRAMplusC4D Consortium. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013;45:25-33.
18. Björkegren JLM, Kovacic JC, Dudley JT, Schadt EE. Genome-wide significant loci: how important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J Am Coll Cardiol 2015;65:830-45.
19. JPatron J, Serra-Cayuela A, Han B, Li C, Wishart DS. Assessing the performance of genome-wide association studies for predicting disease risk. PLoS One 2019;14:e0220215.
21. Libby P. Inflammation in atherosclerosis. Nature 2002;420:868-74.
22. Libby P, Ridker PM, Hansson GK; Leducq Transatlantic Network on Atherothrombosis. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol 2009;54:2129-38.
23. Lowe GD. The relationship between infection, inflammation, and cardiovascular disease: an overview. Ann Periodontol 2001;6:1-8.
24. Libby P, Ridker PM. Inflammation and atherosclerosis: role of C-reactive protein in risk assessment. Am J Med 2004;116:9S-16S.
25. Elkind MS. Infectious burden: a new risk factor and treatment target for atherosclerosis. Infect Disord Drug Targets 2010;10:84-90.
26. Elkind MS, Luna JM, Moon YP, Boden-Albala B, Liu KM, et al. Infectious burden and carotid plaque thickness: the Northern Manhattan Study. Stroke 2010;41:e117-22.
27. Elkind MSV, Ramakrishnan P, Moon YP, Boden-Albala B, Liu KM, et al. Infectious burden and risk of stroke: the Northern Manhattan Study. Arch Neurol 2010;67:33-8.
28. Katan M, Moon YP, Paik MC, Sacco RL, Wright CB, et al. Infectious burden and cognitive function: the Northern Manhattan Study. Neurology 2013;80:1209-15.
29. Desvarieux M, Demmer RT, Rundek T, Boden-Albala B, Jacobs DR Jr, et al; Oral Infections and Vascular Disease Epidemiology Study (INVEST). Relationship between periodontal disease, tooth loss, and carotid artery plaque: the Oral Infections and Vascular Disease Epidemiology Study (INVEST). Stroke 2003;34:2120-5.
30. Desvarieux M, Demmer RT, Rundek T, Boden-Albala B, Jacobs DR Jr, et al. Periodontal microbiota and carotid intima-media thickness: the Oral Infections and Vascular Disease Epidemiology Study (INVEST). Circulation 2005;111:576-82.
31. Engebretson SP, Lamster IB, Elkind MS, Rundek T, Serman NJ, et al. Radiographic measures of chronic periodontitis and carotid artery plaque. Stroke 2005;36:561-6.
32. Desvarieux M, Demmer RT, Jacobs DR Jr, Rundek T, Boden-Albala B, et al. Periodontal bacteria and hypertension: the oral infections and vascular disease epidemiology study (INVEST). J Hypertens 2010;28:1413-21.
33. Boillot A, Demmer RT, Mallat Z, Sacco RL, Jacobs DR, et al. Periodontal microbiota and phospholipases: the Oral Infections and Vascular Disease Epidemiology Study (INVEST). Atherosclerosis 2015;242:418-23.
34. Elter JR, Champagne CM, Offenbacher S, Beck JD. Relationship of periodontal disease and tooth loss to prevalence of coronary heart disease. J Periodontol 2004;75:782-90.
35. Rydén L, Buhlin K, Ekstrand E, de Faire U, Gustafsson A, et al. Periodontitis increases the risk of a first myocardial infarction: a report from the PAROKRANK Study. Circulation 2016;133:576-83.
36. Górski B, Nargiełło E, Grabowska E, Opolski G, Górska R. The association between dental status and risk of acute myocardial infarction among poles: case-control study. Adv Clin Exp Med 2016;25:861-70.
37. Nordendahl E, Gustafsson A, Norhammar A, Näsman P, Rydén L, et al; PAROKRANK Steering Committee. Severe periodontitis is associated with myocardial infarction in females. J Dent Res 2018;97:1114-21.
38. Pasqualini D, Bergandi L, Palumbo L, Borraccino A, Dambra V, et al. Association among oral health, apical periodontitis, CD14 polymorphisms, and coronary heart disease in middle-aged adults. J Endod 2012;38:1570-7.
39. Morita T, Yamazaki Y, Fujiharu C, Ishii T, Seto M, et al. Association between the duration of periodontitis and increased cardiometabolic risk factors: a 9-year cohort study. Metab Syndr Relat Disord 2016;14:475-82.
40. Jönsson D, Orho-Melander M, Demmer RT, Engström G, Melander O, et al. Periodontal disease is associated with carotid plaque area: the Malmö Offspring Dental Study (MODS). J Intern Med 2020;287:301-9.
41. Cotti E, Dessì C, Piras A, Mercuro G. Can a chronic dental infection be considered a cause of cardiovascular disease? A review of the literature. Int J Cardiol 2011;148:4-10.
42. Cotti E, Mercuro G. Apical periodontitis and cardiovascular diseases: previous findings and ongoing research. Int Endod J 2015;48:926-32.
43. Chauhan N, Mittal S, Tewari S, Sen J, Laller K. Association of apical periodontitis with cardiovascular disease via noninvasive assessment of endothelial function and subclinical atherosclerosis. J Endod 2019;45:681-90.
44. Pussinen PJ, Mattila K. Periodontal infections and atherosclerosis: mere associations? Curr Opin Lipidol 2004;15:583-8.
45. Amar S, Wu SC, Madan M. Is Porphyromonas gingivalis cell invasion required for atherogenesis? Pharmacotherapeutic implications. J Immunol 2009;182:1584-92.
46. Epstein SE, Zhu J, Najafi AH, Burnett MS. Insights into the role of infection in atherogenesis and in plaque rupture. Circulation 2009;119:3133-41.
47. Genco R, Offenbacher S, Beck J. Periodontal disease and cardiovascular disease: epidemiology and possible mechanisms. J Am Dent Assoc 2002;133:14S-22.
48. Beck JD, Eke P, Heiss G, Madianos P, Couper D, et al. Periodontal disease and coronary heart disease: a reappraisal of the exposure. Circulation 2005;112:19-24.
49. Beck JD, Moss KL, Morelli T, Offenbacher S. Periodontal profile class is associated with prevalent diabetes, coronary heart disease, stroke, and systemic markers of C-reactive protein and interleukin-6. J Periodontol 2018;89:157-65.
50. Pussinen PJ, Jousilahti P, Alfthan G, Palosuo T, Asikainen S, et al. Antibodies to periodontal pathogens are associated with coronary heart disease. Arterioscler Thromb Vasc Biol 2003;23:1250-4.
51. Pussinen PJ, Alfthan G, Rissanen H, Reunanen A, Asikainen S, et al. Antibodies to periodontal pathogens and stroke risk. Stroke 2004;35:2020-3.
52. Pussinen PJ, Alfthan G, Jousilahti P, Paju S, Tuomilehto J. Systemic exposure to Porphyromonas gingivalis predicts incident stroke. Atherosclerosis 2007;193:222-8.
53. Pussinen PJ, Jauhiainen M, Vilkuna-Rautiainen T, Sundvall J, Vesanen M, et al. Periodontitis decreases the antiatherogenic potency of high density lipoprotein. J Lipid Res 2004;45:139-47.
54. Kim HJ, Cha GS, Kim HJ, Kwon EY, Lee JY, et al. Porphyromonas gingivalis accelerates atherosclerosis through oxidation of high-density lipoprotein. J Periodontal Implant Sci 2018;48:60-8.
55. Pussinen PJ, Könönen E, Paju S, Hyvärinen K, Gursoy UK, et al. Periodontal pathogen carriage, rather than periodontitis, determines the serum antibody levels. J Clin Periodontol 2011;38:405-11.
56. Miyashita H, Honda T, Maekawa T, Takahashi N, Aoki Y, et al. Relationship between serum antibody titres to Porphyromonas gingivalis and hs-CRP levels as inflammatory markers of periodontitis. Arch Oral Biol 2012;57:820-9.
57. Yamazaki K, Honda T, Domon H, Okui T, Kajita K, et al. Relationship of periodontal infection to serum antibody levels to periodontopathic bacteria and inflammatory markers in periodontitis patients with coronary heart disease. Clin Exp Immunol 2007;149:445-52.
58. Sanchez-Torres D, Gutierrez-Bejarano D, Hurtado-Roca Y, Guallar-Castillon P, Muntner P, et al. Non-linear association of periodontal pathogen antibodies with mortality. Int J Cardiol 2015;187:628-36.
59. Pussinen PJ, Paju S, Koponen J, Viikari JSA, Taittonen L, et al. Association of childhood oral infections with cardiovascular risk factors and subclinical atherosclerosis in adulthood. JAMA Netw Open 2019;2:e192523.
60. Akhi R, Wang C, Nissinen AE, Kankaanpää J, Bloigu R, et al. Salivary IgA to MAA-LDL and oral pathogens are linked to coronary disease. J Dent Res 2019;98:296-303.
61. Liljestrand JM, Paju S, Pietiäinen M, Buhlin K, Persson GR, et al. Immunologic burden links periodontitis to acute coronary syndrome. Atherosclerosis 2018;268:177-84.
62. Silver JG, Martin AW, McBride BC. Experimental transient bacteraemias in human subjects with varying degrees of plaque accumulation and gingival inflammation. J Clin Periodontol 1977;4:92-9.
63. Lockhart PB. An analysis of bacteremias during dental extractions. A double-blind, placebo-controlled study of chlorhexidine. Arch Intern Med 1996;156:513-20.
64. Lockhart PB, Brennan MT, Kent ML, Norton HJ, Weinrib DA. Impact of amoxicillin prophylaxis on the incidence, nature, and duration of bacteremia in children after intubation and dental procedures. Circulation 2004;109:2878-84.
65. Tomás I, Diz P, Tobías A, Scully C, Donos N. Periodontal health status and bacteraemia from daily oral activities: systematic review/meta-analysis. J Clin Periodontol 2012;39:213-28.
66. Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of periodontal pathogens in atheromatous plaques. J Periodontol 2000;71:1554-60.
67. Kozarov E, Sweier D, Shelburne C, Progulske-Fox A, Lopatin D. Detection of bacterial DNA in atheromatous plaques by quantitative PCR. Microbes Infect 2006;8:687-93.
68. Toyofuku T, Inoue Y, Kurihara N, Kudo T, Jibiki M, et al. Differential detection rate of periodontopathic bacteria in atherosclerosis. Surg Today 2011;41:1395-400.
69. Pyysalo MJ, Pyysalo LM, Pessi T, Karhunen PJ, Öhman JE. The connection between ruptured cerebral aneurysms and odontogenic bacteria. J Neurol Neurosurg Psychiatry 2013;84:1214-8.
70. Pyysalo MJ, Pyysalo LM, Pessi T, Karhunen PJ, Lehtimäki T, et al. Bacterial DNA findings in ruptured and unruptured intracranial aneurysms. Acta Odontol Scand 2016;74:315-20.
71. Gaetti-Jardim E Jr, Marcelino SL, Feitosa AC, Romito GA, Avila-Campos MJ. Quantitative detection of periodontopathic bacteria in atherosclerotic plaques from coronary arteries. J Med Microbiol 2009;58:1568-75.
72. Calandrini CA, Ribeiro AC, Gonnelli AC, Ota-Tsuzuki C, Rangel LP, et al. Microbial composition of atherosclerotic plaques. Oral Dis 2014;20:e128-34.
73. Szulc M, Kustrzycki W, Janczak D, Michalowska D, Baczynska D, et al. Presence of periodontopathic bacteria DNA in atheromatous plaques from coronary and carotid arteries. Biomed Res Int 2015;2015:825397.
74. Kozarov EV, Dorn BR, Shelburne CE, Dunn WA Jr, Progulske-Fox A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol 2005;25:e17-8.
75. Rafferty B, Jönsson D, Kalachikov S, Demmer RT, Nowygrod R, et al. Impact of monocytic cells on recovery of uncultivable bacteria from atherosclerotic lesions. J Intern Med 2011;270:273-80.
76. Rafferty B, Dolgilevich S, Kalachikov S, Morozova I, Ju J, et al. Cultivation of Enterobacter hormaechei from human atherosclerotic tissue. J Atheroscler Thromb 2011;18:72-81.
77. Cross KL, Campbell JH, Balachandran M, Campbell AG, Cooper SJ, et al. Targeted isolation and cultivation of uncultivated bacteria by reverse genomics. Nat Biotechnol 2019;37:1314-21.
78. Weise H, Naros A, Weise C, Reinert S, Hoefert S. Severe odontogenic infections with septic progress - a constant and increasing challenge: a retrospective analysis. BMC Oral Health 2019;19:173.
79. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 2019;5:eaau3333.
80. Lalla E, Lamster IB, Hofmann MA, Bucciarelli L, Jerud AP, et al. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol 2003;23:1405-11.
81. Ezzahiri R, Stassen FR, Kurvers HA, van Pul MM, Kitslaar PJ, et al. Chlamydia pneumoniae infection induces an unstable atherosclerotic plaque phenotype in LDL-receptor, ApoE double knockout mice. Eur J Vasc Endovasc Surg 2003;26:88-95.
82. Koren O, Spor A, Felin J, Fåk F, Stombaugh J, et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A 2011;108:4592-8.
83. Roth GA, Moser B, Huang SJ, Brandt JS, Huang Y, et al. Infection with a periodontal pathogen induces procoagulant effects in human aortic endothelial cells. J Thromb Haemost 2006;4:2256-61.
84. Gibson FC, Hong C, Chou HH, Yumoto H, Chen J, et al. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation 2004;109:2801-6.
85. Liberto MC, Matera G, Lamberti AG, Barreca GS, Quirino A, et al. In vitro Bartonella quintana infection modulates the programmed cell death and inflammatory reaction of endothelial cells. Diagn Microbiol Infect Dis 2003;45:107-15.
86. Potempa J, Banbula A, Travis J. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol 2000 2000;24:153-92.
87. Potempa J, Sroka A, Imamura T, Travis J. Gingipains, the major cysteine proteinases and virulence factors of Porphyromonas gingivalis: structure, function and assembly of multidomain protein complexes. Curr Protein Pept Sci 2003;4:397-407.
88. Travis J, Banbula A, Potempa J. The role of bacterial and host proteinases in periodontal disease. Adv Exp Med Biol 2000;477:455-65.
89. Johansson A, Kalfas S. Characterization of the proteinase-dependent cytotoxicity of Porphyromonas gingivalis. Eur J Oral Sci 1998;106:863-71.
90. Chen T, Nakayama K, Belliveau L, Duncan MJ. Porphyromonas gingivalis gingipains and adhesion to epithelial cells. Infect Immun 2001;69:3048-56.
91. Roth GA, Ankersmit HJ, Brown VB, Papapanou PN, Schmidt AM, et al. Porphyromonas gingivalis infection and cell death in human aortic endothelial cells. FEMS Microbiol Lett 2007;272:106-13.
92. Hirasawa M, Kurita-Ochiai T. Porphyromonas gingivalis induces apoptosis and autophagy via ER stress in human umbilical vein endothelial cells. Mediators Inflamm 2018;2018:1967506.
93. Pham K, Feik D, Hammond BF, Rams TE, Whitaker EJ. Aggregation of human platelets by gingipain-R from Porphyromonas gingivalis cells and membrane vesicles. Platelets 2002;13:21-30.
94. Lourbakos A, Yuan YP, Jenkins AL, Travis J, Andrade-Gordon P, et al. Activation of protease-activated receptors by gingipains from Porphyromonas gingivalis leads to platelet aggregation: a new trait in microbial pathogenicity. Blood 2001;97:3790-7.
95. Arvanitidis E, Bizzarro S, Alvarez Rodriguez E, Loos BG, Nicu EA. Reduced platelet hyper-reactivity and platelet-leukocyte aggregation after periodontal therapy. Thromb J 2017;15:5.
96. Klarström Engström K, Khalaf H, Kälvegren H, Bengtsson T. The role of Porphyromonas gingivalis gingipains in platelet activation and innate immune modulation. Mol Oral Microbiol 2015;30:62-73.
97. Lönn J, Ljunggren S, Klarström-Engström K, Demirel I, Bengtsson T, et al. Lipoprotein modifications by gingipains of Porphyromonas gingivalis. J Periodontal Res 2018;53:403-13.
98. Joo JY, Cha GS, Chung J, Lee JY, Kim SJ, et al. Peptide 19 of porphyromonas gingivalis heat shock protein is a potent inducer of low-density lipoprotein oxidation. J Periodontol 2017;88:e58-e64.
99. Wallet SM, Puri V, Gibson FC. Linkage of infection to adverse systemic complications: periodontal disease, toll-like receptors, and other pattern recognition systems. Vaccines (Basel) 2018;6:21.
100. Hernández-Ríos P, Pussinen PJ, Vernal R, Hernández M. Oxidative stress in the local and systemic events of apical periodontitis. Front Physiol 2017;8:869.
101. Sessa R, Pietro MD, Filardo S, Turriziani O. Infectious burden and atherosclerosis: a clinical issue. World J Clin Cases 2014;2:240-9.
102. He F, Zuo L. Redox roles of reactive oxygen species in cardiovascular diseases. Int J Mol Sci 2015;16:27770-80.
103. Di Pietro M, Filardo S, Falasca F, Turriziani O, Sessa R. Infectious agents in atherosclerotic cardiovascular diseases through oxidative stress. Int J Mol Sci 2017;18:2459.
104. Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res 2017;120:713-35.
105. Li L, Michel R, Cohen J, Decarlo A, Kozarov E. Intracellular survival and vascular cell-to-cell transmission of Porphyromonas gingivalis. BMC Microbiol 2008;8:26.
106. Azenabor AA, Yang S, Job G, Adedokun OO. Elicitation of reactive oxygen species in Chlamydia pneumoniae-stimulated macrophages: a Ca2+-dependent process involving simultaneous activation of NADPH oxidase and cytochrome oxidase genes. Med Microbiol Immunol 2005;194:91-103.
107. Di Pietro M, Filardo S, De Santis F, Sessa R. Chlamydia pneumoniae infection in atherosclerotic lesion development through oxidative stress: a brief overview. Int J Mol Sci 2013;14:15105-20.
108. Yamaguchi Y, Kurita-Ochiai T, Kobayashi R, Suzuki T, Ando T. Activation of the NLRP3 inflammasome in Porphyromonas gingivalis-accelerated atherosclerosis. Pathog Dis 2015;73:ftv011.
109. Li XY, Wang C, Xiang XR, Chen FC, Yang CM, et al. Porphyromonas gingivalis lipopolysaccharide increases lipid accumulation by affecting CD36 and ATP-binding cassette transporter A1 in macrophages. Oncol Rep 2013;30:1329-36.
110. Pollreisz A, Huang Y, Roth GA, Cheng B, Kebschull M, et al. Enhanced monocyte migration and pro-inflammatory cytokine production by Porphyromonas gingivalis infection. J Periodontal Res 2010;45:239-45.
111. Sheets SM, Potempa J, Travis J, Casiano CA, Fletcher HM. Gingipains from Porphyromonas gingivalis W83 induce cell adhesion molecule cleavage and apoptosis in endothelial cells. Infect Immun 2005;73:1543-52.
112. Bugueno IM, Khelif Y, Seelam N, Morand DN, Tenenbaum H, et al. Porphyromonas gingivalis differentially modulates cell death profile in Ox-LDL and TNF-α pre-treated endothelial cells. PLoS One 2016;11:e0154590.
113. Shiheido Y, Maejima Y, Suzuki JI, Aoyama N, Kaneko M, et al. Porphyromonas gingivalis, a periodontal pathogen, enhances myocardial vulnerability, thereby promoting post-infarct cardiac rupture. J Mol Cell Cardiol 2016;99:123-37.
114. Amberger A, Maczek C, Jürgens G, Michaelis D, Schett G, et al. Co-expression of ICAM-1, VCAM-1, ELAM-1 and Hsp60 in human arterial and venous endothelial cells in response to cytokines and oxidized low-density lipoproteins. Cell Stress Chaperones 1997;2:94-103.
115. Takei A, Huang Y, Lopes-Virella MF. Expression of adhesion molecules by human endothelial cells exposed to oxidized low density lipoprotein. Influences of degree of oxidation and location of oxidized LDL. Atherosclerosis 2001;154:79-86.
116. Malekmohammad K, Sewell RDE, Rafieian-Kopaei M. Antioxidants and atherosclerosis: mechanistic aspects. Biomolecules 2019;9:301.
117. Hulsmans M, Holvoet P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J Cell Mol Med 2010;14:70-8.
118. Li L, Messas E, Batista EL Jr, Levine RA, Amar S. Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model. Circulation 2002;105:861-7.
119. Jain A, Batista EL Jr, Serhan C, Stahl GL, Van Dyke TE. Role for periodontitis in the progression of lipid deposition in an animal model. Infect Immun 2003;71:6012-8.
120. Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res 2004;45:1169-96.
121. Hardardóttir I, Grunfeld C, Feingold KR. Effects of endotoxin on lipid metabolism. Biochem Soc Trans 1995;23:1013-8.
122. Pirillo A, Catapano AL, Norata GD. HDL in infectious diseases and sepsis. Handb Exp Pharmacol 2015;224:483-508.
123. Lassenius MI, Pietiläinen KH, Kaartinen K, Pussinen PJ, Syrjänen J, et al; FinnDiane Study Group. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care 2011;34:1809-15.
124. Feingold KR, Grunfeld C. in Endotext KR, Feingold et al., Eds. MDText.com, Inc., South Dartmouth (MA); 2000. Available from https://www.ncbi.nlm.nih.gov/books/NBK326741/ [Last accessed on 27 Feb 2020].
125. Grunfeld C, Marshall M, Shigenaga JK, Moser AH, Tobias P, et al. Lipoproteins inhibit macrophage activation by lipoteichoic acid. J Lipid Res 1999;40:245-52.
126. Moretti E, Gonnelli S, Campagna M, Nuti R, Collodel G, et al. Influence of Helicobacter pylori infection on metabolic parameters and body composition of dyslipidemic patients. Intern Emerg Med 2014;9:767-72.
127. Saxlin T, Suominen-Taipale L, Kattainen A, Marniemi J, Knuuttila M, et al. Association between serum lipid levels and periodontal infection. J Clin Periodontol 2008;35:1040-7.
128. Korhonen S, Saxlin T, Suominen L, Jula A, Knuuttila M, et al. Serum cholesterol ratios and periodontal infection: results of the Health 2000 Survey. J Clin Periodontol 2011;38:787-94.
129. Nishimura F, Taniguchi A, Yamaguchi-Morimoto M, Soga Y, Iwamoto Y, et al. Periodontal infection and dyslipidemia in type 2 diabetics: association with increased HMG-CoA reductase expression. Horm Metab Res 2006;38:530-5.
130. Nepomuceno R, Pigossi SC, Finoti LS, Orrico SRP, Cirelli JA, et al. Serum lipid levels in patients with periodontal disease: a meta-analysis and meta-regression. J Clin Periodontol 2017;44:1192-207.
131. Van Dyke TE. The management of inflammation in periodontal disease. J Periodontol 2008;79:1601-8.
132. Van Dyke TE, Kornman KS. Inflammation and factors that may regulate inflammatory response. J Periodontol 2008;79:1503-7.
133. Hasturk H, Kantarci A, Van Dyke TE. Paradigm shift in the pharmacological management of periodontal diseases. Front Oral Biol 2012;15:160-76.
134. Grover V, Malhotra R, Kapoor A, Singh J, Sachdeva S. Proresolution mediators and receptors: novel drug targets for enhancing pharmacological armamentarium against periodontal inflammation. Infect Disord Drug Targets 2013;13:75-84.
135. Hasturk H, Abdallah R, Kantarci A, Nguyen D, Giordano N, et al. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler Thromb Vasc Biol 2015;35:1123-33.
136. Hamilton JA, Hasturk H, Kantarci A, Serhan CN, Van Dyke T. Atherosclerosis, periodontal disease, and treatment with resolvins. Curr Atheroscler Rep 2017;19:57.
137. Elter JR, Hinderliter AL, Offenbacher S, Beck JD, Caughey M, et al. The effects of periodontal therapy on vascular endothelial function: a pilot trial. Am Heart J 2006;151:47.
138. Shimada Y, Komatsu Y, Ikezawa-Suzuki I, Tai H, Sugita N, et al. The effect of periodontal treatment on serum leptin, interleukin-6, and C-reactive protein. J Periodontol 2010;81:1118-23.
139. Desvarieux M, Demmer RT, Jacobs DR, Papapanou PN, Sacco RL, et al. Changes in clinical and microbiological periodontal profiles relate to progression of carotid intima-media thickness: the Oral Infections and Vascular Disease Epidemiology study. J Am Heart Assoc 2013;2:e000254.
140. D’Aiuto F, Nibali L, Parkar M, Patel K, Suvan J, et al. Oxidative stress, systemic inflammation, and severe periodontitis. J Dent Res 2010;89:1241-6.
141. D’Aiuto F, Orlandi M, Gunsolley JC. Evidence that periodontal treatment improves biomarkers and CVD outcomes. J Clin Periodontol 2013;40:S85-105.
142. Inchingolo F, Marrelli M, Annibali S, Cristalli MP, Dipalma G, et al. Influence of endodontic treatment on systemic oxidative stress. Int J Med Sci 2013;11:1-6.
143. Saffi MAL, Rabelo-Silva ER, Polanczyk CA, Furtado MV, Montenegro MM, et al. Periodontal therapy and endothelial function in coronary artery disease: a randomized controlled trial. Oral Dis 2018;24:1349-57.
144. Wang Y, Andrukhov O, Rausch-Fan X. Oxidative stress and antioxidant system in periodontitis. Front Physiol 2017;8:910.
145. Scannapieco FA, Dasanayake AP, Chhun N. Does periodontal therapy reduce the risk for systemic diseases?. Dent Clin North Am 2010;54:163-81.
146. Falcao A, Bullón P. A review of the influence of periodontal treatment in systemic diseases. Periodontol 2000 2019;79:117-28.
147. Orlandi M, Suvan J, Petrie A, Donos N, Masi S, et al. Association between periodontal disease and its treatment, flow-mediated dilatation and carotid intima-media thickness: a systematic review and meta-analysis. Atherosclerosis 2014;236:39-46.
148. Jackson LA, Campbell LA, Kuo CC, Rodriguez DI, Lee A, et al. Isolation of Chlamydia pneumoniae from a carotid endarterectomy specimen. J Infect Dis 1997;176:292-5.
149. Kozarov E. Bacterial invasion of vascular cell types: vascular infectology and atherogenesis. Future Cardiol 2012;8:123-38.
150. Libby P. Fanning the flames: inflammation in cardiovascular diseases. Cardiovasc Res 2015;107:307-9.
151. Joensen JB, Juul S, Henneberg E, Thomsen G, Ostergaard L, et al. Can long-term antibiotic treatment prevent progression of peripheral arterial occlusive disease? A large, randomized, double-blinded, placebo-controlled trial. Atherosclerosis 2008;196:937-42.
152. Vainas T, Stassen FR, Schurink GW, Tordoir JH, Welten RJ, et al. Secondary prevention of atherosclerosis through chlamydia pneumoniae eradication (SPACE Trial): a randomised clinical trial in patients with peripheral arterial disease. Eur J Vasc Endovasc Surg 2005;29:403-11.
153. Everett BM, Pradhan AD, Solomon DH, Paynter N, Macfadyen J, et al. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J 2013;166:199-207.e15.
154. Ridker PM. Closing the loop on inflammation and atherothrombosis: why perform the CIRT and CANTOS trials? Trans Am Clin Climatol Assoc 2013;124:174-90.
155. Ridker PM. Moving beyond JUPITER: will inhibiting inflammation reduce vascular event rates? Curr Atheroscler Rep 2013;15:295.
156. Moreira DM, Lueneberg ME, da Silva RL, Fattah T, Mascia Gottschall CA. Rationale and design of the TETHYS trial: the effects of methotrexate therapy on myocardial infarction with ST-segment elevation. Cardiology 2013;126:167-70.
157. O’Donoghue ML, Braunwald E, White HD, Lukas MA, Tarka E, et al. Effect of darapladib on major coronary events after an acute coronary syndrome: the SOLID-TIMI 52 randomized clinical trial. JAMA 2014;312:1006-15.
158. Dunne MW. Rationale and design of a secondary prevention trial of antibiotic use in patients after myocardial infarction: the WIZARD (weekly intervention with zithromax [azithromycin] for atherosclerosis and its related disorders) trial. J Infect Dis 2000;181:S572-8.
159. Libby P, Egan D, Skarlatos S. Roles of infectious agents in atherosclerosis and restenosis: an assessment of the evidence and need for future research. Circulation 1997;96:4095-103.
160. Amar S, Engelke M. Periodontal innate immune mechanisms relevant to atherosclerosis. Mol Oral Microbiol 2015;30:171-85.
161. Kesavalu L, Lucas AR, Verma RK, Liu L, Dai E, et al. Increased atherogenesis during Streptococcus mutans infection in ApoE-null mice. J Dent Res 2012;91:255-60.
162. Kozarov E, Huber K, Wojta J. Infection-associated biomarkers of inflammation in atherosclerosis. Curr Pharm Des 2015;21:1776-82.
163. Kramer CD, Simas AM, He X, Ingalls RR, Weinberg EO, et al. Distinct roles for dietary lipids and Porphyromonas gingivalis infection on atherosclerosis progression and the gut microbiota. Anaerobe 2017;45:19-30.
164. Anderson JL. Infection, antibiotics, and atherothrombosis--end of the road or new beginnings? N Engl J Med 2005;352:1706-9.
165. Gieffers J, Füllgraf H, Jahn J, Klinger M, Dalhoff K, et al. Chlamydia pneumoniae infection in circulating human monocytes is refractory to antibiotic treatment. Circulation 2001;103:351-6.
166. Deshpande RG, Khan M, Genco CA. Invasion strategies of the oral pathogen porphyromonas gingivalis: implications for cardiovascular disease. Invasion Metastasis ;18:57-69.
167. Dorn BR, Dunn WA Jr, Progulske-Fox A. Invasion of human coronary artery cells by periodontal pathogens. Infect Immun 1999;67:5792-8.
168. Lamont RJ, Chan A, Belton CM, Izutsu KT, Vasel D, et al. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun 1995;63:3878-85.
169. Progulske-Fox A, Kozarov E, Dorn B, Dunn W Jr, Burks J, et al. Porphyromonas gingivalis virulence factors and invasion of cells of the cardiovascular system. J Periodontal Res 1999;34:393-9.
170. Wunsch CM, Lewis JP. Porphyromonas gingivalis as a Model Organism for Assessing Interaction of Anaerobic Bacteria with Host Cells. J Vis Exp 2015:e53408.
171. Griffen AL, Becker MR, Lyons SR, Moeschberger ML, Leys EJ. Prevalence of Porphyromonas gingivalis and periodontal health status. J Clin Microbiol 1998;36:3239-42.
172. Rudney JD, Chen R. The vital status of human buccal epithelial cells and the bacteria associated with them. Arch Oral Biol 2006;51:291-8.
173. Donley TG. Time to put periodontal disease on the list of chronic inflammatory diseases contributing to premature atherosclerosis. CMAJ 2019;191:E52.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Kozarov E, Progulske-Fox A. Atherosclerosis microbiome: upcoming target for vaccine and drug development. Vessel Plus 2020;4:10. http://dx.doi.org/10.20517/2574-1209.2019.31
AMA Style
Kozarov E, Progulske-Fox A. Atherosclerosis microbiome: upcoming target for vaccine and drug development. Vessel Plus. 2020; 4: 10. http://dx.doi.org/10.20517/2574-1209.2019.31
Chicago/Turabian Style
Kozarov, Emil, Ann Progulske-Fox. 2020. "Atherosclerosis microbiome: upcoming target for vaccine and drug development" Vessel Plus. 4: 10. http://dx.doi.org/10.20517/2574-1209.2019.31
ACS Style
Kozarov, E.; Progulske-Fox A. Atherosclerosis microbiome: upcoming target for vaccine and drug development. Vessel Plus. 2020, 4, 10. http://dx.doi.org/10.20517/2574-1209.2019.31
About This Article
Copyright

Data & Comments
Data


 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 15
likes
Like This Article 15
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.