
Vascular smooth muscle cell contractile function and mechanotransduction


Abstract
Vascular smooth muscle cells (VSMCs) are the predominant cell type in the arterial wall and normally adopt a quiescent, contractile phenotype to regulate vascular tone. In the arterial wall, VSMCs are exposed to multiple mechanical cues, including stretch and matrix stiffness, which regulate VSMC contraction. However, during ageing and in vascular disease, such as atherosclerosis, hypertension and vascular calcification, the arterial wall stiffens and VSMC contraction contributes to this process. VSMCs display remarkable plasticity and changes in their mechanical environment promote VSMCs to adopt a proliferative, synthetic phenotype. VSMC phenotypic modulation is associated with altered expression of contractile proteins that generate actomyosin-based force. However, our understanding of precise mechanisms whereby altered mechanical landscape and mechanotransduction influence VSMC contraction remains limited. In this review, we discuss the present literature describing how VSMCs sense and respond to changes in their mechanical environment and how these changes influence VSMC contraction.
Keywords
Introduction
Cardiovascular disease (CVD) remains one of most prevalent risk factors to health worldwide, and is the second highest cause of mortality within the UK[1]. The aberration of health caused from CVD places a heavy burden on the health-care of developing countries as well as representing a major cause of death and morbidity in industrialised countries[2,3]. CVD is an umbrella term, which holds host to multiple related diseases, including peripheral arterial disease, coronary heart disease and stroke[4]. The risk factors of each vary depending on the specificity of the disease, however many present common symptoms, providing a crucial link in treatment methods. Common physiological risk factors are hypertension, obesity, a rise in cholesterol and diabetes with treatment of each providing a larger scale preventative therapy of CVD[5]. Despite this, the largest risk factor associated with CVD is ageing, an inevitable process that all individuals undergo.
Ageing causes an increase in CVD incidence and prevalence due to wide array of changes that occurs as a person becomes older. The cause of ageing requires a diverse and intricate investigation into the crosstalk between multiple genetic and environmental (i.e., diet, exercise and smoking) factors[6]. A key modification observed is the structural and functional alterations within the vasculature. This includes the stiffening of the arteries, in particular the aorta. The function of the aorta is crucial in converting the large output of oxygenated blood from the left ventricle into a more controlled flow within the smaller arterioles and capillaries[7]. Stiffening ultimately reduces aortic compliance and increases systolic arterial pressure that augments the overall vascular resistance[8]. To compensate, the left ventricle adopts a compensatory mechanism creating a change in the end-systolic volume as well as prolonging systolic contraction. A direct consequence of this is the thickening of the left ventricle, which causes an aberrant hypertrophic physiology[9]. Coupled to this, ageing also causes defects in the repair mechanisms of the vasculature which further drives the diseased-phenotypic changes[10]. This, in turn, diminishes the capability of the vascular system to overcome the increased workload that is generated as a repercussion. Thus, ageing presents as the most predicative cause of CVD.
Arterial stiffness: cause and relevance in CVD
Arterial stiffness is a predicative biomarker in ageing and CVD, including atherosclerosis, hypertension and obese populations[11-13]. Normally, pulse pressure expands the elastic arteries, transferring energy from the blood to the arterial wall and slowing pulse velocity. In conditions of enhanced arterial stiffness, pulse pressure is no longer able to expand the artery, increasing pulse velocity, and pulse pressure is transmitted to the microcirculation of organs such as the heart and lungs[14]. Vessels of the microcirculation are more fragile, resulting in damage to the microcirculation. The current gold standard method in assessing arterial stiffness is pulse wave velocimetry (PWV)[15]. A higher PWV is linked to individuals who have greater risk of CVD[16]. As a result, this method can be utilised to provide a predictive analysis of CVD independent of standard blood pressure measurements of the brachial artery[17].
Arterial structure
Elastic arteries, including the aorta, are structurally composed of three layers; the tunica intima is the innermost layer [Figure 1]. It is comprised of a sheet of endothelial cells along with a basal membrane and collagen fibrils. The tunica media neighbours the tunica intima as the middle layer and is primarily composed of vascular smooth muscle cells (VSMCs) reinforced with elastin and collagen fibrils. The final and outermost layer is the tunica adventitia, containing largely connective tissue as a means to provide reinforcement to the structure of the aorta[18]. This allows the aorta to act as an “elastic buffering chamber” in order to store and transmit blood to the peripheral circulation during systole and diastole, respectively[19].
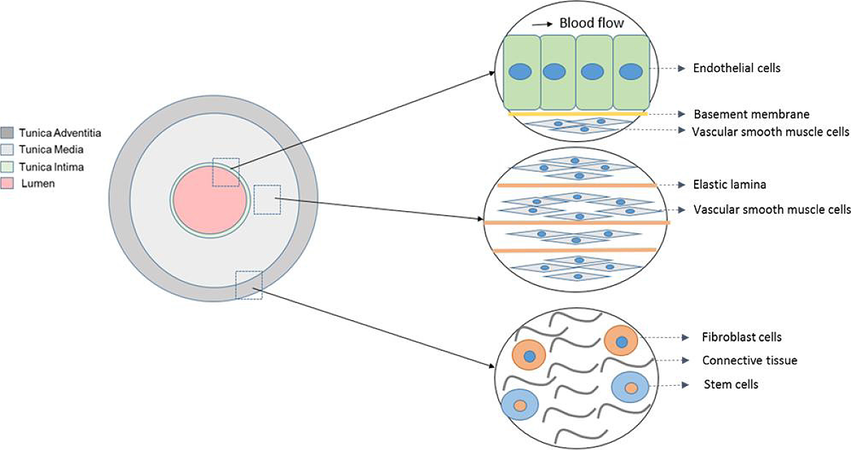
Figure 1. Schematic representation of arterial structural organisation
This function becomes aberrant when aortic stiffness increases. Stiffness augmentation of the aorta is ultimately driven by changes in extracellular matrix (ECM) composition, in particular enhanced elastic degradation as well as augmented collagen deposition[20,21]. All three arterial layers are mechanoresponsive and remodel during vascular disease progression[22-24]. In this review, we focus on the response of the VSMC layer to enhanced matrix stiffness. Elastin in small arteries and the aorta allows for vascular distensibility and in normal conditions, is found in high abundance within these vessels[21]. However, this dogmatic view on arterial stiffness was recently found to be misleading, as an in depth analysis of the published data on hypertension found collagen levels to be inconsistent[25]. Due to this, several studies have now found multiple contributing factors towards aortic stiffness, ranging from, but not limited to, mechanical stimuli, inflammatory cytokines and compositional changes in the ECM[25]. The existing treatment therapies of aortic stiffness involve different anti-hypertensive drugs, which have an indirect effect on arterial stiffness[25]. These drugs block calcium-channels and angiotensin II receptors, both of which modulate VSMC contraction and vascular tone, as opposed to ECM composition. Therefore, VSMCs have a fundamental role in the aortic stiffness and much of the focus has now shifted onto VSMC structure and function[26].
VSMC phenotypic modulation and the cytoskeleton
VSMC are the predominant cell type within the arterial wall. They are arranged in a fibrous helix and regulate vessel diameter and vascular tone[27]. Within a mature artery, VSMCs exist in a quiescent, contractile state and regulate vascular tone via vessel constriction[28]. However, VSMCs retain phenotypic plasticity and can dedifferentiate into a proliferative, synthetic state[29,30]. VSMC phenotypic modulation is associated with developmental and disease associated vessel remodeling, where VSMCs exhibit higher rates of proliferation, migration and altered ECM deposition[29,30].
The key filamentous components of the VSMC cytoskeleton are the intermediate filaments, microtubules and actin. Intermediate filaments, including vimentin and desmin, maintain VSMCs 3D structure[31]. In contrast, the properties of microtubules are not as clearly defined due to variable tissue types and staining methods. Actin filaments transmit mechanical signals to dense plaques which act as signalling hubs and are found dispersed within the cytoplasm[24]. Three different isoforms of actin exist, alpha, beta and gamma actin, with alpha actin being the abundant isoform typical within contractile VSMCs[24,32]. Changes in both extracellular and intracellular tension, alter actin cytoskeletal organisation and regulate cell contraction, migration and survival[33].
VSMC phenotypic modulation is commonly associated with altered contractile marker expression[34]. Contractile VSMCs possess smooth muscle myosin II (SM-myosin II), smoothelin and smooth muscle-actin and these are downregulated in models where arterial injury[35]. SM-myosin II is the dominant myosin isoform found within contractile VSMC and is composed of both two heavy and light chains. There are two different types of light chains identified as myosin light chain-20 (MLC-20) and MLC-17, with the phosphorylation of the former regulating VSMC contraction[36]. In contrast, synthetic VSMCs contain non-muscle myosin II (NM-myosin II), which is upregulated in the proliferative state[35]. NM-myosin II is also expressed in the differentiated state, albeit at lower levels. Once phosphorylated, myosin associates with actin filaments to form the actomyosin complex. The ATPase activity of the myosin then results in rapid cycling of the cross-bridges formed between actin and myosin, thus causing a pulling of the actin thin filaments[36]. The physiological function of the two myosin isoforms vary and the SM-myosin II has a higher immune-reactivity in tissue areas where faster phasic VSMC contractions were occurring[37]. This is in contrast to NM-myosin II, which primarily regulates the slower tonic phase of VSMC contraction[37]. The tonic contraction induced from NM-myosin II generates less force than phasic contraction produced from SM-myosin II[37,38].
VSMC contraction
Calcium-dependent pathway
VSMC contraction occurs via two interlinked pathways that contribute synergistically to the contractile properties of VSMCs [Figure 2]. The first pathway, more commonly known as the calcium-dependent pathway, primarily involves augmenting cytoplasmic calcium levels to induce phasic contraction. Increased intracellular calcium can be triggered by mechanical, electrical and chemical stimuli, either by calcium influx from channels located on the plasma membrane or by release of calcium from the sarcoplasmic reticulum (SR)[39]. Calcium entry from the extracellular space usually occurs via voltage gated calcium channels (VGCCs) or non-selective cation channels. Sub-populations of L-type, P/Q-type and T-type VGCCs are all found within VSMCs and are activated via depolarization[40]. In addition, non-selective cation channels, found to predominantly be members of the transient receptor potential canonical family, allow for Na+ and Ca2+ influx following receptor occupancy or capicitative calcium entry[40].
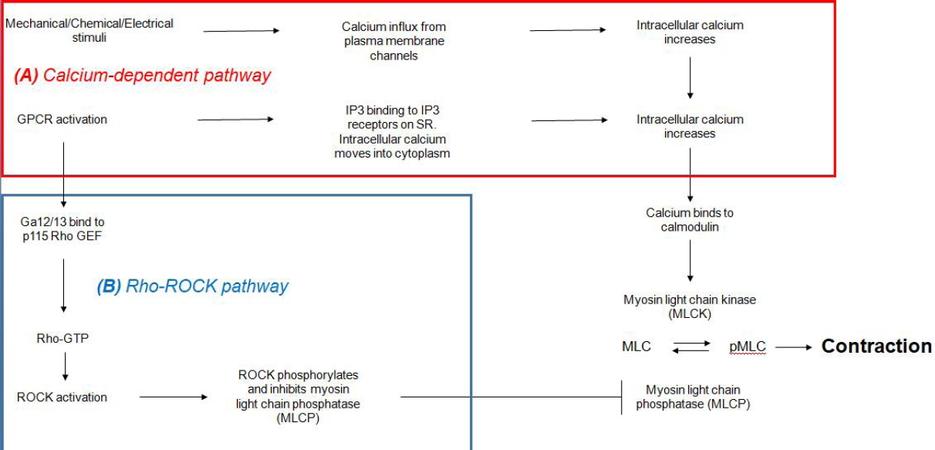
Figure 2. Calcium dependent and independent regulation of VSMC contraction. The two pathways work synergistically. Calcium dependent regulation is associated with transient phasic contraction whereas RhoA/ROCK regulation is associated with the prolonged tonic contraction of VSMCs. GPCR: G-protein coupled receptor; IP3: inositol triphosphate; SR: sarcoplasmic reticulum; Rho GEF: RhoGTPase guanine nucleotide exchange factors; ROCK: Rho-associated protein kinase; MLC: myosin light chain; VSMC: vascular smooth muscle cell
Release of calcium from the SR is predominantly mediated by the activation of G-protein coupled receptors (GPCRs) (i.e., the AT1 receptor) coupled to the Gaq G-protein. The Gaq protein, when in its GTP-bound state, causes activation of phospholipase C which hydrolyses phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol triphosphate (IP3) and diacylglycerol[41,42]. IP3 binds to the IP3 receptors present on the sarcolemma, which causes opening of the calcium channels and subsequent depletion of calcium intracellular store.
After an increase of cytoplasmic calcium concentration has been established, calmodulin becomes bound by 4 calcium ions. The resulting calcium-calmodulin complex interacts with and activates myosin light chain kinase (MLCK)[43]. Next, MLCK phosphorylates MLC-20 (also known as the regulatory light chain) on the serine-19 and threonine-18 residues. The phosphorylation of serine-19 causes a resulting increase in the activity of the Mg2+-ATPase and this effect is further enhanced by the phosphorylation of the latter residue. From this, the cross-bridge cycling is initiated and the myosin head can actively pull on the thin filament of the actin to induce contraction of the muscle[44].
Rho/Rho-associated protein kinase pathway
In the absence of external contractile stimuli, the MLC-20 light chain remains phosphorylated at a low level. This low level leads to a slower tonic form of contraction, which regulates the vascular tone[36]. A calcium-independent pathway that involves Rho/Rho-associated protein kinase (ROCK) signalling regulates VSMC tonic contractions [Figure 2]. This pathway not only caters to contractile function, but also extends to smooth muscle cell migration, proliferation and apoptosis[45]. RhoA, part of the Ras superfamily, is a GTPase which can act as a molecular switch between a GTP/GDP bound state[46]. In resting conditions, the Rho GDP dissociation inhibitor targets GDP-Rho for binding, as a means to localise the GTPase from the membrane to the cytosol. However, activation of GPCR receptors, in particular Ga12/13 subtypes, can catalyse GTP for GDP exchange in RhoA by binding to p115 RhoGTPase guanine nucleotide exchange factors[47]. In its GTP bound form, RhoA can interact with target proteins by utilising its C-terminal geranyl-geranylated tail to anchor itself to the plasma membrane[47].
One of the target proteins activated by RhoA is ROCK[48]. ROCK is a member of the protein kinase A, G and C family of protein kinases, and is characterised as a serine/threonine kinase. There are two isoforms of this kinase, referred to as ROCK1 and ROCK2, with expression of both present in VSMCs[49]. Its structure is composed of an N-terminal kinase domain, a central coiled-coil domain and a C-terminal pleckstrin homology domain that associates with the Rho GTPase[47]. ROCK has many effects within VSMCs and influences actomyosin activity by two main pathways. Firstly, ROCK actively regulates cytoskeletal organisation by preventing actin filament depolymerisation[48]. Secondly, ROCK inhibits myosin light chain phosphatase (MLCP). MLCP has a structure that is composed of three subunits; a 37 kDa catalytic subunit, a variable subunit and a myosin-binding subunit[36]. The myosin-binding site is crucial for its regulation and is subject to phosphorylation, specifically at residues. Threonine-695/697 (major site), serine-849/854 and threonine-850/855[47,50]. Phosphorylation prevents MLCP from regulating the MLC phosphorylation state and increases the basal phosphorylated MLC level, stimulating VSMC contraction and augmenting vascular tone[50].
Membrane anchors to the actin cytoskeleton
VSMCs make connective junctions to their surrounding environment, which includes the ECM and neighboring cells within the vasculature. These adhesions play a vital role in determining morphology and VSMC function. The adhesion molecules that are utilised by VSMCs can be separated, despite their structural and functional similarities[51].
Cell-cell adhesions
Cadherins are the primary molecules in cell-cell adhesion formation. The most abundant isoforms are E (epithelial)-, P (placental)- and N (neuronal)-cadherins, all of which belong to the type I classical cadherin family[52,53]. N-cadherin is the predominant cadherin in VSMCs and mediates cell-cell adhesion formation with neighbouring endothelial cells as well as other VSMCs[53]. The N-cadherin adhesion plays important roles in regulating VSMC function. N-cadherin adhesions suppress both VSMC proliferation and apoptosis[54,55]. N-cadherin is also involved in VSMC migration; however, its exact role currently remains unclarified[56].
N-cadherin is composed of a large extracellular N-terminal domain, flanked by a single trans-membrane anchoring domain and a small cytoplasmic tail[52]. The large extracellular domain consists of five cadherin (EC) repeat regions that are important in coupling N-cadherin into a parallel homodimer via linkage between adjacent EC repeats[52,53]. EC repeats require calcium binding which stabilises the interaction between the parallel cadherin molecules[56,57]. The cadherin homodimer of one cell interacts with the homodimer of an adjacent cell by interchanging a specific beta strand, referred to as the A* strand, found in the EC1 domain[58]. This interaction, referred to as trans-binding, is necessary for the formation of cell-cell adhesions[53]. The cytoplasmic tail of N-cadherin is linked to the actin cytoskeleton via a number of cytoskeleton-associated proteins, including β-catenin, p120-catenin and α-catenin. α-catenin is recruited to the adhesion structure via β-catenin and plays a crucial role in providing a link between the N-cadherin-based junction and the actin cytoskeleton[59].
Cadherin-based mechanotransduction is observed in multiple cell types and induces adhesion and cytoskeletal remodelling, altered adhesion strength and changes in actomyosin activity. Evidence suggests that N-cadherin-based adhesions are important for VSMC contraction and mechanotransduction; β-catenin recruitment to N-cadherin-based adhesions is necessary for VSMC contraction, and N-cadherin is essential for VSMC myogenic response to changes in pressure. Despite this evidence, our understanding of the role of N-cadherin-based mechanotransduction in VSMC function remains poorly defined.
Cell-matrix adhesions
Cell-matrix adhesions possess integrin receptors at their core[60]. Integrins span the plasma membrane and physically associate with different ECM components[61]. Integrin receptors perform both structural and mechanosensing signalling functions within cell-matrix adhesions. Integrins form heterodimers, consisting of an alpha and beta subunit[62] and are structurally comprised of an extracellular ligand-binding domain, which binds the ECM, and a cytosolic domain, which is anchored to the actin cytoskeleton[63]. Before transducing intracellular tension to the ECM, the integrin receptors must mature via the recruitment of further integrin receptors as well as other cytoskeletal components[64]. Talin and alpha-actinin bind directly to the cytosolic domain of integrin and talin binding promotes the recruitment of additional components to cell matrix adhesions, including vinculin, paxillin and focal adhesion kinase[62,65]. Vinculin consists of 3-stuctural regions known as the head, neck and tail domain[66]. The vinculin binding site is auto-inhibited by interactions between its head and tail domain[66]. This interaction is disrupted by talin/alpha actinin, via individual binding or cooperatively with PIP2[67]. Once disrupted, the activated form can then associate with cell-matrix adhesions via talin[67]. Physical stress induces exposure of vinculin binding sites on talin’s rod domain allowing vinculin binding. In addition, the vinculin molecule also associates with actin filaments, thereby crosslinking the actin cytoskeleton to the integrin receptors. This allows force transduction from the contractile machinery of VSMCs to be transmitted to the ECM[66].
The linker of nucleoskeleton and cytoskeleton complex
The nuclear envelope (NE) is a double lipid bilayer that consists of an outer nuclear membrane (ONM) and an inner nuclear membrane (INM), separated by a perinuclear space [Figure 3]. The ONM and INM are continuous and join at nuclear pores[68]. A meshwork of A-type and B-type lamin intermediate proteins and lamina associated proteins, collectively known as the nuclear lamina, underlies the INM and provides structural support to the NE[68]. The linker of nucleoskeleton and cytoskeleton (LINC) complex, comprised of nesprin-family members and SUN-domain containing proteins, spans the NE. Giant nesprin-1/2 isoforms reside on the ONM and associate with filamentous actin via N-terminal calponin homology (CH) domains[69]. Within the perinuclear space, binding of the nesprin Klarsicht, Anc-1, Syne-1 homology-domain to the SUN-domain of SUN1/2 maintains LINC complex stability[70,71]. At the INM, SUN1/2 interact with lamins A/C[71], allowing the plasma membrane and nucleus to function as a mechanically coupled system [Figure 3].
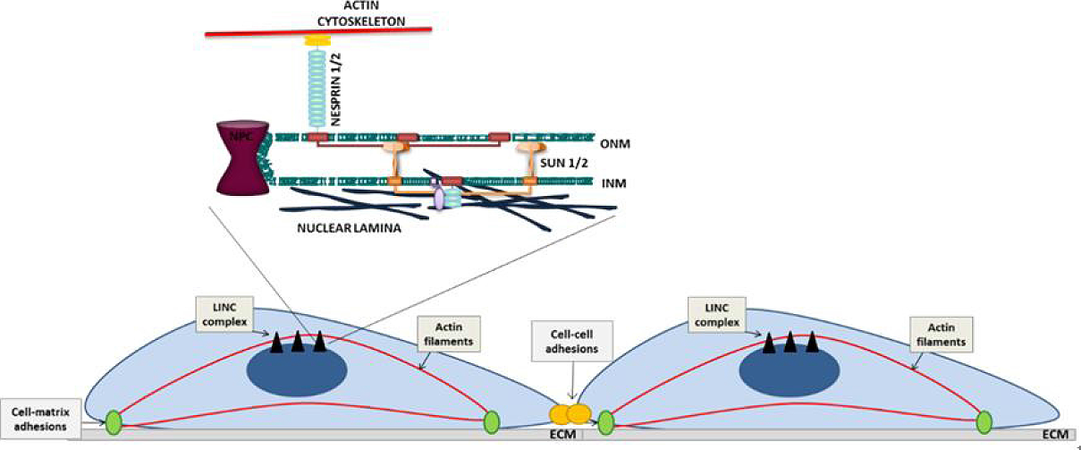
Figure 3. Schematic representation of adhesion organisation and the LINC complex in VSMCs. ONM: outer nuclear membrane; INM: inner nuclear membrane; ECM: extracellular matrix; LINC: linker of nucleoskeleton and cytoskeleton; VSMC: vascular smooth muscle cell
The LINC complex is a regulator of cytoskeletal organisation and directly transmits biophysical signals into the nucleus. The LINC complex is subjected to mechanical tension and actomyosin-generated forces are directly transduced across the NE to the nuclear lamina[72,73]. However, the LINC complex exists in mechanical balance with mechanotransduction and LINC disruption triggers cell-matrix adhesion, cell-cell adhesion and cytoskeletal reorganisation[74,75]. LINC complex disruption also alters actomyosin activity, nesprin disruption enhances actomyosin activity in skeletal muscle progenitor and endothelial cells, whereas lamin A disruption in skeletal muscle progenitor cells enhanced actomyosin activity and reduced actomyosin activity in fibroblast cells[76,77]. This suggests that the LINC complex plays cell-specific roles in regulating actomyosin activity and disruption of the nuclear lamin A during ageing alters VSMC morphology and cell-matrix organisation[78]. However, the role of the LINC complex in VSMC actomyosin regulation remains unknown. Although the mechanism of this LINC complex/actomyosin feedback remains unknown, lamin A/C and SUN2 regulate Rac1 and RhoA activity, respectively[78,79].
Mechanotransduction and the ECM
Sensing the extracellular environment
Extracellular mechanical cues directly regulate VSMC function, including actomyosin activity, adhesion, differentiation and migration[66]. To achieve this, VSMCs must convert mechanical signals into biochemical response via a process known as mechanotransduction. Mechanosensors range from stretch-sensitive channels, cytoskeletal filaments, cytosolic proteins and nuclear proteins, all of which undergo conformational changes when encountering intra/extracellular tension[80].
Conformational changes induced by tension alters mechanosensors modification, interactions and localisation within the cell[81]. Vinculin, in particular, acts as a regulator of mechanical stress in addition to its role as a mechano-coupler. As a result, cells regulate their function by actively exerting and resisting forces both to and from the ECM as a means to adjust their mechanical properties[82]. Force is transmitted via cell-matrix adhesions, which serve as bidirectional signalling conduits, enabling “inside-out” and “outside-in” signalling[82]. This is crucial for the maintenance of normal physiology as well as injury-repair, by offsetting signalling pathways that induce appropriate gene expression. Due to this, the cell can enable structural remodelling of the cytoskeleton, which allows for correct adjustment of the vascular tone and in turn the blood flow[24].
Mechanotransduction: role of stretch
The cyclical process of the cardiac pumping creates numerous mechanical stimuli, all of which the vascular smooth muscle is exposed to. Examples are transmural pressure, circumferential wall tension and vascular shear strain[24]. Shear stress predominantly acts on the tunica intima, where endothelial cells reside. In the tunica media, VSMCs are subjected to cyclic stretch that originates from the pulsatile blood pressure[27]. Pulsatile stretch is cyclical in nature due to a rise in blood flow during the systolic phase and then a gradual decrease when in the diastolic phase[83]. This form of pressure induces rapid cell-matrix and actin filament reorganisation[24]. Stretch also physically opens mechanically gated cation channels, promoting Ca2+ ion entry and VSMC contraction[84,85]. Stretch signals also regulate activity of several important signalling molecules, including protein kinase C and Akt. Therefore stretch signals regulate a range of VSMC functions, including proliferation, migration and apoptosis[27].
CVD and the role of matrix stiffness
VSMCs can sense the stiffness of their surrounding matrix and respond by exerting actomyosin-generated tension on the ECM. As the stiffness of a material increases, its elasticity decreases[86]. Therefore, as matrix stiffness is augmented, vessel stretch signals decrease and there is a switch from transient stretch signals to sustained stiffness signalling. Decreased arterial compliance is commonly found in the early stages of numerous CVDs such as atherosclerosis, restenosis and aneurism. Atherosclerosis is an inflammatory CVD that acts as the underlying cause of heart attack, stroke and cardiac death[87]. The development of this disease primarily involves the endothelial intimal layer and the vascular smooth muscle medial layer[88] and results in the formation of an atherosclerotic plaque, containing a lipid core surrounded by a fibrous cap. Atherosclerosis decreases aortic compliance and the stiffness of the individual components of an atherosclerotic plaque has been measured by atomic force microscopy, which displays a range of stiffness, from the soft lipid core (~5 kPa), to the relatively healthy cellular regions (~10 kPa), to the stiff fibrous cap (60-250 kPa)[89].
VSMC phenotypic modulation, proliferation and migration is prevalent in atherosclerosis and there is a clear coloration between changes in arterial compliance and atherosclerotic disease progression[11,90]. Yet our understanding of the influence of matrix stiffness on VSMC function remains limited. VSMCs exposed to enhanced matrix stiffness display increased intracellular tension and form larger cell-matrix adhesions[25,91]. VSMC stiffness is also augmented in hypertension and diabetes[25,26]. Importantly, treatment with actin inhibitors reduce VSMCs stiffness, suggesting that the stiffer aortic environments increase actomyosin force generation in VSMCs[26].
In addition to altered actomyosin activity, matrix stiffness also potentially influences VSMC differentiation. VSMC phenotype is regulated by numerous environmental cues, including the ECM[92]. Atherosclerotic plaques display reduced levels of elastin and collagen-I accumulation[93]. Fetal aortic VSMCs display enhanced traction forces and actomyosin activity when matrix stiffness is increased from 10 kPa to 25 kPa[93]. However, they fail to produce sufficient force to displace a 135 kPa matrix[93], suggesting that VSMCs lose the ability to contract and deform their surrounding ECM under enhanced matrix stiffness. In agreement with this notion, VSMCs lose the ability to contract and deform the ECM when surrounded by stiff collagen-I fibrils in 3D models[94]. In stiff environments, VSMCs exert higher levels of mechanical tension, which directs a proliferative change to the synthetic phenotype[94]. Expression levels of key contractile marker proteins, such as alpha smooth muscle actin, are reduced when collagen-I gel concentration is increased[92]. Furthermore, VSMCs transition to the synthetic state when stiffness and nanotopography of the substrate is increased[95]. Therefore, ECM stiffness influences VSMC phenotypic modulation.
As described above, matrix stiffness influences both the expression of VSMC contractile proteins and the ability of VSMCs to physically contract and deform their surrounding ECM. Therefore, stiffened ECM is no longer remodeled by the intrinsic actomyosin activity of the VSMCs[96]. However, this does not explain whether the absence of substrate deformation is due to a decline in VSMC actomyosin response or because the ECM has become too stiff to be manipulated. For example, a recent study investigated the ex-vivo vasoconstrictor response of young and old soleus muscle feed arteries. Despite an upregulation of ROCK activity, there was a decreased constrictor response in the aged vessels[96]. Aged VSMCs were incapable of generating sufficient force to induce matrix remodeling[96]. However, ECM deformation was used to assess the vasoconstrictor response and given that aged arteries are stiffer than their younger counterparts, VSMC actomyosin activity may remain intact[97]. In agreement with this, cell-matrix adhesions were subjected to increased mechanical load in aged arteries, compared to their younger counterparts[96]. Further investigation is now required to assess whether the mechanisms of VSMC actomyosin signaling is influenced by matrix stiffness.
Finally, directional cellular migration is also induced by gradients of ECM stiffness, known as durotaxis. Durotaxis contrasts from chemotaxis and haptotaxis due to an absence of soluble chemical signals or adhesive ligand density, respectively[98]. Both healthy and diseased tissues possess heterogeneity in their mechanical stiffness indicating the presence of gradients[89,99-102]. VSMC directional migration is prevalent in atherosclerosis and VSMCs are exposed to low (~5 kPa) and high matrix stiffness (> 200 kPa) within the atherosclerotic plaque. VSMCs orientate in the direction of an ECM stiffness gradient[98] and show a directed migration towards a mechanical gradient when plated on gels coated with fibronectin as opposed to laminin[103]. Fibronectin is found in high concentrations in atherosclerotic lesions, suggesting that ECM composition is a key component of VMSC durotaxis. It remains unknown whether durotaxis, chemotaxis and haptotaxis work cooperatively to regulate VSMC migration in atherosclerosis, however, matrix stiffness gradients may participate in the enhanced VSMC migration observed in vascular diseases such as atherosclerosis.
Conclusion
Evidence clearly dictates that matrix stiffness plays a crucial role in CVD. However, its effects on VSMCs, the predominant cell type within the aorta, remains poorly defined. The majority of VSMC research has been performed on tissue culture plastic or glass, which are over 1000 times stiffer than an arterial wall. There is a pressing need to utilise materials that more closely replicate both physiological and pathological stiffness in VSMC research. Several important questions remained unanswered: (1) the signalling pathways regulating VSMC function in response to matrix stiffness remain to be fully elucidated; (2) do VSMCs from different vascular beds possess unique force generating capabilities in response to matrix stiffness; and (3) how do other cell types, including endothelial cells, tune VSMC actomyosin activity in response to matrix stiffness? Answering these questions will facilitate an understanding of the aetiology of arterial stiffness on VSMC function that will potentially allow development of new therapeutic avenues for the treatment of a wide range of CVDs.
Declarations
Authors’ contributionsResponsible for the design, literature review, writing and editing of this manuscript: Ahmed S, Warren DT
Availability of data and materialsNot applicable.
Financial support and sponsorshipA British Heart Foundation (BHF) Non-Clinical PhD Studentship (FS/17/32/32916) funded this work.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Bhatnagar P, Wickramasinghe K, Wilkins E, Townsend N. Trends in the epidemiology of cardiovascular disease in the UK. Heart 2016;102:1945-52.
2. Islam AM, Mohibullah A, Paul T. Cardiovascular disease in Bangladesh: a review. Bangladesh Heart J 2016;31:80-99.
3. Ness AR, Powles JW. Fruit and vegetables, and cardiovascular disease: a review. Int J Epidemiol 1997;26:1-13.
4. Stewart J, Manmathan G, Wilkinson P. Primary prevention of cardiovascular disease: a review of contemporary guidance and literature. JRSM Cardiovasc Dis 2017;6:2048004016687211.
5. Sun LY, Lee EW, Zahra A, Park JH. Risk factors of cardiovascular disease and their related socio-economical, environmental and health behavioral factors: focused on low-middle income countries-a narrative review article. Iran J Public Health 2015;44:435-44.
8. Cecelja M, Chowienczyk P. Role of arterial stiffness in cardiovascular disease. JRSM Cardiovasc Dis 2012; doi: 10.1258/cvd.2012.012016.
9. Strait JB, Lakatta EG. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail Clin 2012;8:143-64.
10. Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci 2010;65:1028-41.
11. Palombo C, Kozakova M. Arterial stiffness, atherosclerosis and cardiovascular risk: pathophysiologic mechanisms and emerging clinical indications. Vascul Pharmacol 2016;77:1-7.
13. Safar ME, Czernichow S, Blacher J. Obesity, arterial stiffness, and cardiovascular risk. J Am Soc Nephrol 2006;17:S109-11.
14. Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, et al. Arterial stiffness and cardiovascular events: the Framingham heart study. Circulation 2010;121:505-11.
15. Sethi S, Rivera O, Oliveros R, Chilton R. Aortic stiffness: pathophysiology, clinical implications, and approach to treatment. Integr Blood Press Control 2014;7:29-34.
17. DeLoach SS, Townsend RR. Vascular stiffness: its measurement and significance for epidemiologic and outcome studies. Clin J Am Soc Nephrol 2008;3:184-92.
18. Tsamis A, Krawiec JT, Vorp DA. Elastin and collagen fibre microstructure of the human aorta in ageing and disease: a review. J R Soc Interface 2013;10:20121004.
19. Belz GG. Elastic properties and windkessel function of the human aorta. Cardiovasc Drugs Ther 1995;9:73-83.
20. Steed MM, Tyagi N, Sen U, Schuschke DA, Joshua IG, et al. Functional consequences of the collagen/elastin switch in vascular remodeling in hyperhomocysteinemic wild-type, eNOS-/-, and iNOS-/- mice. Am J Physiol Lung Cell Mol Physiol 2010;299:L301-11.
21. Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev 2009;89:957-89.
22. Jufri NF, Mohamedali A, Avolio A, Baker MS. Mechanical stretch: physiological and pathological implications for human vascular endothelial cells. Vasc Cell 2015;7:8.
23. Humphrey JD. Mechanisms of arterial remodeling in hypertension: coupled roles of wall shear and intramural stress. Hypertension 2008;52:195-200.
24. Ye GJ, Nesmith AP, Parker KK. The role of mechanotransduction on vascular smooth muscle myocytes cytoskeleton and contractile function. Anat Rec (Hoboken) 2014;297:1758-69.
25. Sehgel NL, Vatner SF, Meininger GA. “Smooth muscle cell stiffness syndrome”-revisiting the structural basis of arterial stiffness. Front Physiol 2015;6:335.
26. Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, et al. Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res 2010;107:615-9.
27. Haga JH, Li YS, Chien S. Molecular basis of the effects of mechanical stretch on vascular smooth muscle cells. J Biomech 2007;40:947-60.
28. Brozovich FV, Nicholson CJ, Degen CV, Gao YZ, Aggarwal M, et al. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacol Rev 2016;68:476-532.
29. Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol 2012;74:13-40.
30. Owens GK. Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. Novartis Found Symp 2007;283:174-91.
31. Yamin R, Morgan KG. Deciphering actin cytoskeletal function in the contractile vascular smooth muscle cell. J Physiol 2012;590:4145-54.
32. Skalli O, Ropraz P, Trzeciak A, Benzonana G, Gillessen D, et al. A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol 1986;103:2787-96.
33. Papakonstanti EA, Stournaras C. Cell responses regulated by early reorganization of actin cytoskeleton. FEBS Lett 2008;582:2120-7.
34. Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg 2007;45:A25-32.
35. Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J 2007;15:100-8.
37. Eddinger TJ, Meer DP. Myosin II isoforms in smooth muscle: heterogeneity and function. Am J Physiol Cell Physiol 2007;293:C493-508.
38. Löfgren M, Ekblad E, Morano I, Arner A. Nonmuscle myosin motor of smooth muscle. J Gen Physiol 2003;121:301-10.
39. Woodrum DA, Brophy CM. The paradox of smooth muscle physiology. Mol Cell Endocrinol 2001;177:135-43.
40. Martinsen A, Dessy C, Morel N. Regulation of calcium channels in smooth muscle: new insights into the role of myosin light chain kinase. Channels (Austin) 2014;8:402-13.
41. Fridlyand LE, Philipson LH. Pancreatic beta cell G-protein coupled receptors and second messenger interactions: a systems biology computational analysis. PloS One 2016;11:e0152869.
42. Inagami T, Eguchi S, Tsuzuki S, Ichiki T. Angiotensin II receptors AT1 and AT2: new mechanisms of signaling and antagonistic effects of AT1 and AT2. In: Dhalla NS, Zahradka P, Dixon IMC, Beamish RE, editors. Angiotensin II receptor blockade physiological and clinical implications. Boston: Springer; 1998. pp. 129-39.
43. Walsh MP. Calmodulin and its roles in skeletal muscle function. Can Anaesth Soc J 1983;30:390-8.
44. Lee S, Kumar S. Actomyosin stress fiber mechanosensing in 2D and 3D. F1000Res 2016; doi: 10.12688/f1000research.8800.1.
45. Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol 2010;2:a005066.
47. Fukata Y, Amano M, Kaibuchi K. Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol Sci 2001;22:32-9.
48. Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 2010;67:545-54.
50. Murányi A, Derkach D, Erdodi F, Kiss A, Ito M, et al. Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett 2005;579:6611-5.
51. Burute M, Thery M. Spatial segregation between cell-cell and cell-matrix adhesions. Curr Opin Cell Biol 2012;24:628-36.
52. Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, et al. Cell-cell adhesion and communication. Molecular cell biology. New York: W. H. Freeman; 2000.
53. Sun Z, Parrish AR, Hill MA, Meininger GA. N-cadherin, a vascular smooth muscle cell-cell adhesion molecule: function and signaling for vasomotor control. Microcirculation 2014;21:208-18.
54. Lyon CA, Johnson JL, White S, Sala-Newby GB, George SJ. EC4, a truncation of soluble N-cadherin, reduces vascular smooth muscle cell apoptosis and markers of atherosclerotic plaque instability. Mol Ther Methods Clin Dev 2014;1:14004.
55. Lyon CA, Wadey KS, George SJ. Soluble N-cadherin: a novel inhibitor of VSMC proliferation and intimal thickening. Vascul Pharmacol 2016;78:53-62.
56. Lyon CA, Koutsouki E, Aguilera CM, Blaschuk OW, George SJ. Inhibition of N-cadherin retards smooth muscle cell migration and intimal thickening via induction of apoptosis. J Vasc Surg 2010;52:1301-9.
57. Perez TD, Nelson WJ. Cadherin adhesion: mechanisms and molecular interactions. Handb Exp Pharmacol 2004; doi: 10.1007/978-3-540-68170-0_1.
58. Shapiro L, Weis WI. Structure and biochemistry of cadherins and catenins. Cold Spring Harb Perspect Biol 2009;1:a003053.
59. Weis WI, Nelson WJ. Re-solving the cadherin-catenin-actin conundrum. J Biol Chem 2006;281:35593-7.
60. Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta 2004;1692:103-19.
62. Suki B, Parameswaran H, Imsirovic J, Bartolák-Suki E. Regulatory roles of fluctuation-driven mechanotransduction in cell function. Physiology (Bethesda) 2016;31:346-58.
63. Bachir AI, Zareno J, Moissoglu K, Plow EF, Gratton E, et al. Integrin-associated complexes form hierarchically with variable stoichiometry in nascent adhesions. Curr Biol 2014;24:1845-53.
64. Romer LH, Birukov KG, Garcia JG. Focal adhesions: paradigm for a signaling nexus. Circ Res 2006;98:606-16.
65. Chorev DS, Volberg T, Livne A, Eisenstein M, Martins B, et al. Conformational states during vinculin unlocking differentially regulate focal adhesion properties. Sci Rep 2018;8:2693.
66. Mierke CT. The role of vinculin in the regulation of the mechanical properties of cells. Cell Biochem Biophys 2009;53:115-26.
67. Carisey A, Ballestrem C. Vinculin, an adapter protein in control of cell adhesion signalling. Eur J Cell Biol 2011;90:157-63.
68. Burke B, Ellenberg J. Remodelling the walls of the nucleus. Nat Rev Mol Cell Biol 2002;3:487-97.
69. Warren DT, Zhang Q, Weissberg PL, Shanahan CM. Nesprins: intracellular scaffolds that maintain cell architecture and coordinate cell function? Expert Rev Mol Med 2005;7:1-15.
70. Haque F, Lloyd DJ, Smallwood DT, Dent CL, Shanahan CM, et al. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol 2006;26:3738-51.
71. Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, et al. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 2006;172:41-53.
72. Guilluy C, Osborne LD, Van Landeghem L, Sharek L, Superfine R, et al. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat Cell Biol 2014;16:376-81.
73. Lombardi ML, Jaalouk DE, Shanahan CM, Burke B, Roux KJ, et al. The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J Biol Chem 2011;286:26743-53.
74. Stewart RM, Zubek AE, Rosowski KA, Schreiner SM, Horsley V, et al. Nuclear-cytoskeletal linkages facilitate cross talk between the nucleus and intercellular adhesions. J Cell Biol 2015;209:403-18.
75. Chambliss AB, Khatau SB, Erdenberger N, Robinson DK, Hodzic D, et al. The LINC-anchored actin cap connects the extracellular milieu to the nucleus for ultrafast mechanotransduction. Sci Rep 2013;3:1087.
76. Schwartz C, Fischer M, Mamchaoui K, Bigot A, Lok T, et al. Lamins and nesprin-1 mediate inside-out mechanical coupling in muscle cell precursors through FHOD1. Sci Rep 2017;7:1253.
77. Chancellor TJ, Lee J, Thodeti CK, Lele T. Actomyosin tension exerted on the nucleus through nesprin-1 connections influences endothelial cell adhesion, migration, and cyclic strain-induced reorientation. Biophys J 2010;99:115-23.
78. Porter LJ, Holt MR, Soong D, Shanahan CM, Warren DT. Prelamin a accumulation attenuates rac1 activity and increases the intrinsic migrational persistence of aged vascular smooth muscle cells. Cells 2016; doi: 10.3390/cells5040041.
79. Thakar K, May CK, Rogers A, Carroll CW. Opposing roles for distinct LINC complexes in regulation of the small GTPase RhoA. Mol Biol Cell 2017;28:182-91.
80. Alonso JL, Goldmann WH. Cellular mechanotransduction. AIMS Biophys 2016;3:50-62.
81. Belaadi N, Aureille J, Guilluy C. Under pressure: mechanical stress management in the nucleus. Cells 2016; doi: 10.3390/cells5020027.
82. Wrighton KH. Cell adhesion: the ‘ins’ and ‘outs’ of integrin signalling. Nat Rev Mol Cell Biol 2013;14:752.
83. Anwar MA, Shalhoub J, Lim CS, Gohel MS, Davies AH. The effect of pressure-induced mechanical stretch on vascular wall differential gene expression. J Vasc Res 2012;49:463-78.
84. Ducret T, El Arrouchi J, Courtois A, Quignard JF, Marthan R, et al. Stretch-activated channels in pulmonary arterial smooth muscle cells from normoxic and chronically hypoxic rats. Cell calcium 2010;48:251-9.
85. Zou H, Lifshitz LM, Tuft RA, Fogarty KE, Singer JJ. Visualization of Ca2+ entry through single stretch-activated cation channels. Proc Natl Acad Sci U S A 2002;99:6404-9.
86. Humphrey JD, Harrison DG, Figueroa CA, Lacolley P, Laurent S. Central artery stiffness in hypertension and aging: a problem with cause and consequence. Circ Res 2016;118:379-81.
87. Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol 2015;209:13-22.
88. Kher N, Marsh JD. Pathobiology of atherosclerosis--a brief review. Semin Thromb Hemost 2004;30:665-72.
89. Tracqui P, Broisat A, Toczek J, Mesnier N, Ohayon J, et al. Mapping elasticity moduli of atherosclerotic plaque in situ via atomic force microscopy. J Struct Biol 2011;174:115-23.
90. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res 2016;118:692-702.
91. Hytönen VP, Wehrle-Haller B. Mechanosensing in cell-matrix adhesions - converting tension into chemical signals. Exp Cell Res 2016;343:35-41.
92. Timraz SBH, Rezgui R, Boularaoui SM, Teo JCM. Stiffness of extracellular matrix components modulates the phenotype of human smooth muscle cells in vitro and allows for the control of properties of engineered tissues. Procedia Eng 2015;110:29-36.
93. Sazonova OV, Isenberg BC, Herrmann J, Lee KL, Purwada A, et al. Extracellular matrix presentation modulates vascular smooth muscle cell mechanotransduction. Matrix Biol 2015;41:36-43.
94. McDaniel DP, Shaw GA, Elliott JT, Bhadriraju K, Meuse C, et al. The stiffness of collagen fibrils influences vascular smooth muscle cell phenotype. Biophys J 2007;92:1759-69.
95. Chaterji S, Kim P, Choe SH, Tsui JH, Lam CH, et al. Synergistic effects of matrix nanotopography and stiffness on vascular smooth muscle cell function. Tissue Eng Part A 2014;20:2115-26.
96. Seawright JW, Sreenivasappa H, Gibbs HC, Padgham S, Shin SY, et al. Vascular smooth muscle contractile function declines with age in skeletal muscle feed arteries. Front Physiol 2018;9:856.
97. Kohn JC, Lampi MC, Reinhart-King CA. Age-related vascular stiffening: causes and consequences. Front Genet 2015;6:112.
98. Isenberg BC, Dimilla PA, Walker M, Kim S, Wong JY. Vascular smooth muscle cell durotaxis depends on substrate stiffness gradient strength. Biophys J 2009;97:1313-22.
99. Berry MF, Engler AJ, Woo YJ, Pirolli TJ, Bish LT, et al. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am J Physiol Heart Circ Physiol 2006;290:H2196-203.
100. Liu F, Tschumperlin DJ. Micro-mechanical characterization of lung tissue using atomic force microscopy. J Vis Exp 2011; doi: 10.3791/2911.
101. Lopez JI, Kang I, You WK, McDonald DM, Weaver VM. In situ force mapping of mammary gland transformation. Integr Biol (Camb) 2011;3:910-21.
102. Plodinec M, Loparic M, Monnier CA, Obermann EC, Zanetti-Dallenbach R, et al. The nanomechanical signature of breast cancer. Nat Nanotechnol 2012;7:757-65.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ahmed S, Warren DT. Vascular smooth muscle cell contractile function and mechanotransduction. Vessel Plus 2018;2:36. http://dx.doi.org/10.20517/2574-1209.2018.51
AMA Style
Ahmed S, Warren DT. Vascular smooth muscle cell contractile function and mechanotransduction. Vessel Plus. 2018; 2: 36. http://dx.doi.org/10.20517/2574-1209.2018.51
Chicago/Turabian Style
Ahmed, Sultan, Derek T. Warren. 2018. "Vascular smooth muscle cell contractile function and mechanotransduction" Vessel Plus. 2: 36. http://dx.doi.org/10.20517/2574-1209.2018.51
ACS Style
Ahmed, S.; Warren DT. Vascular smooth muscle cell contractile function and mechanotransduction. Vessel Plus. 2018, 2, 36. http://dx.doi.org/10.20517/2574-1209.2018.51
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 49 clicks
Cite This Article 49 clicks

 Like This Article 5
likes
Like This Article 5
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.