
Thrombospondins and remodeling of the tumor microenvironment


Abstract
Vascular remodeling defines cancer growth and aggressiveness. Although cancer cells produce pro-angiogenic signals, the fate of angiogenesis critically depends on the cancer microenvironment. Composition of the extracellular matrix (ECM) and tumor inflammation determine whether a cancer will remain dormant, will be recognized by the immune system and eliminated, or whether the tumor will develop and lead to the spread and metastasis of cancer cells. Thrombospondins (TSPs), a family of ECM proteins that has long been associated with the regulation of angiogenesis and cancer, regulate multiple physiological processes that determine cancer growth and spreading, from angiogenesis to inflammation, metabolic changes, and properties of ECM. Here, we sought to review publications that describe various functions of TSPs that link these proteins to regulation of cancer growth by modulating multiple physiological and pathological events that prevent or support tumor development. In addition to its direct effects on angiogenesis, TSPs have important roles in regulation of inflammation, immunity, ECM properties and composition, and glucose and insulin metabolism. Furthermore, TSPs have distinct roles as regulators of remodeling in tissues and tumors, such that the pathways activated by a single TSP can interact and influence each other. The complex nature of TSP interactions and functions, including their different cell- and tissue-specific effects, may lead to confusing results and controversial conclusions when taken out of the context of interdisciplinary and holistic approaches. However, studies of TSP functions and roles in different systems of the organism offer an integrative view of tumor remodeling and a potential for finding therapeutic targets that would modulate multiple complementary processes associated with cancer growth.
Keywords
Introduction
The studies of cancer initiation and progression have focused on the molecular and cellular signaling events that lead to changes in cancer cell differentiation, proliferation, and apoptosis, all of which initiate uncontrolled growth and spreading. The roles of immunity and of the response of the whole organism to cancer cells have been historically appreciated, but the role of the microenvironment in the initiation, progression, and spreading of cancer has become a more active field of study only recently. It has been accepted that cancer cells are constantly forming in a healthy body[1,2] but they do not survive. They may remain dormant for years due to the healthy microenvironment, which does not support the tumor growth. The tumor microenvironment, which prevents or promotes cancer growth, consists of stromal and vascular cells, immune and inflammatory cells. Additionally, the extracellular matrix (ECM) and secreted signals that these cells generate promote or restrict cancer cell division and migration[3,4]. The progression of cancer depends on the complex interplay between the tumor cells and the tumor microenvironment. Targeting the components of the tumor microenvironment is now a recognized powerful tool of cancer therapy and prevention of spreading and recurrence.
The development of tumor vasculature depends on angiogenesis, accompanied by inflammation, which is an important factor in predicting tumor vascularization, growth, and spreading. Targeting the tumor vasculature has been an active approach in finding new therapeutic targets for many years[5] but, unfortunately, has not fulfilled the expectations of cancer therapies due to significant side effects and adverse events in tumors in response to hypoxia[6]. It has become clear that we have a limited appreciation of pathological processes associated with the tumor microenvironment and, as a result, inadequate understanding of potential therapies that may improve the microenvironment and restrict tumor growth.
Tumor angiogenesis and the recruitment of immune and inflammatory cells into the tumor rely on the composition of ECM[7]. One important event that occurs during tumor progression is the stiffening of the ECM, caused by the deposition of collagen and fibronectin, leading to increased proliferation and tumor advancement[8]. Cancer-associated fibroblasts are important contributors of ECM stiffening[9]. Tumor ECM is also modified by vascular and blood cells that release proteases and chemoattractants and deposit ECM to promote angiogenesis, additional recruitment of vascular and inflammatory cells, and inflammation[10].
Tumor inflammation is closely associated with the tumor aggressiveness and metastasis[11]. Activated cancer and vascular cells produce chemoattractants and pro-inflammatory signals to recruit inflammatory cells from blood. The accumulation of inflammatory cells in a tumor is an important prognostic index that has been successfully used to evaluate the aggressiveness of cancer in conjunction with other indexes that describe the proliferation rate of cancer cells and their migratory potential. CD68, a marker of macrophages is one of the 16 markers evaluated in Oncotype DX, a clinical test that is used to make therapeutic decisions and predict the aggressiveness of breast cancer[12,13]. The recruitment and retention of inflammatory cells in tumors depends on the ECM composition[14].
This article reviews the contribution of thrombospondins (TSPs), a family of secreted ECM proteins, in regulation of the cancer microenvironment and the initiation and progression of tumor growth that is defined by the vascular and ECM remodeling and inflammatory response. It is becoming clear that TSPs affect every pathological process associated with cancer advancement and are key protein regulators of the tissue remodeling that occurs with cancer growth.
TSPs and cancer
The ECM is complex and ever-changing. All the cells in a tumor constantly remodel the ECM and deposit growth factors, proteases, pro-inflammatory and chemoattractant proteins into ECM. The composition of ECM defines whether it will support cancer growth, angiogenesis, and inflammation by providing specific growth- and migration-promoting signals and changing the physical properties of the tissue. ECM contains structural components, e.g., collagens, and secreted proteins that define the interactions of the cells with the structural elements of the ECM. One of the influential groups of proteins that regulates the interactions between structural proteins and cells are TSPs. This family of proteins consists of five members in humans (TSP-1 - TSP-5). The five members of the family share a high level of homology and a number of properties[15]. However, they have unique domains and protein sequences that render each of the five TSPs distinct in their interactions with the ligands in ECM and with cell surface receptors. As a result, TSPs have distinct functions in tissue remodeling and regulation of cancer growth[16]. Furthermore, a strong and repeated association of TSP expression or suppression in various cancers highlights their role in cancer regulation[17].
TSP-3, TSP-4, and TSP-5
TSPs of group B (TSP-3, TSP-4, and TSP-5) are evolutionarily older proteins with fewer domains than in the group A TSP proteins[18]. Group B TSPs are important in embryonic development[16,19] and participate in the activation of embryonic tissue remodeling programs[20]. TSP-3 and TSP-5 are involved in the regulation of growth plate organization and limb length[21]. Complete deletion of TSP-3 and TSP-5 leads to reduced limb length, which signifies their prominent role in skeletal growth[21]. Not surprisingly, TSP-3 is also linked to cancer angiogenesis, metastasis and invasion in osteosarcoma patients[22].
TSP-4 is one of the highly upregulated genes (in the top 1%) in several types of cancer, e.g., gastric cancer[23-25] and breast cancer[26-28]. Its expression is upregulated in stromal tissue of invasive breast and gastric adenoma cancers[20,29]. A recent study suggests that the loss of miR-142, resulting in high expression of TSP-4, enhances hepatocellular carcinoma (HCC) invasion and progression. Therefore, targeting TSP-4 may be an important strategy to treat HCC[30]. Increased expression of TSP-4 in ECM promotes invasion of the breast cancer cells[20]. Another study stated that TSP-4 mRNA expression in fibroblasts was stimulated by cancer cells, suggesting that TSP-4 is an important novel marker in the detection of diffuse-type gastric adenocarcinomas[29]. Flexible heteroarotinoid compounds coordinate growth, apoptosis and differentiation of cancer cells. One of the compounds of this group, SHetA2, inhibits angiogenic effects by decreasing the secretion of TSP-4, along with vascular endothelial growth factor A and fibroblast growth factor, in ovarian and renal cancers[31].
TSP-4 promotes cancer angiogenesis and growth in mouse models of breast cancer[32]. Knocking out TSP-4 in mice resulted in smaller tumors with decreased numbers of endothelial cells and lower levels of angiogenesis markers. Conversely, a P387 variant of TSP-4 that is a more active variant of TSP-4 in cellular effects and interactions with ligands[32], had increased cancer angiogenesis and tumor growth[32]. Although the vascular cells appear to be the main source of TSP-4 in breast cancers[32,33], in vivo, the cancer cells themselves also produce small amounts of TSP-4 that appear to be sufficient to support angiogenesis and cancer growth even in TSP-4 deficient animals[32,33]. Complete deletion of TSP-4, in both the host and the cancer cells, is required in order to document effects on tumor growth[33]. In addition to these effects in tumors, TSP-4 promotes adhesion and migration of leukocytes[34]. Thus, TSP-4 is a pro-angiogenic[32] and a pro-inflammatory protein[35] that supports tumor growth by activating multiple complementary pathways.
TSP-4 mediates the effects of transforming growth factor beta (TGF-β), a master regulator of ECM and inflammation, on angiogenesis[33]. The direct roles of TSP-4 in ECM regulation remain poorly understood. However, it is clear that TSP-4 regulates collagen production and can prevent fibrosis in tissues[36].
On the other hand, TSP-4 serves as a tumor suppressor in colorectal cancer and suppresses in vitro tumor colony formation[37]. Epigenetic profiling studies revealed that hypermethylation of the TSP-4 promoter leads to its inactivation and loss of TSP-4 tumor suppressor function in cutaneous T cell lymphoma[38]. The opposite effects of TSPs on cancer cells and on the cancer microenvironment are a recurrent theme when studying the roles of TSPs in cancer regulation, leading to controversial findings that are difficult to explain. Ultimately, the results from in vivo studies, where TSPs levels have been manipulated, should be considered to help define physiological roles for individual TSPs in cancer regulation.
TSP-1 and TSP-2
The more recently developed group of TSPs, which includes TSP-1 and TSP-2 and is termed “group A”, appeared in evolution along with the development of the cardiovascular system[19,39]. These two proteins have several C-terminal domains homologous to group B proteins but differ from group B TSPs in their N-terminal protein parts[40]. The newly acquired in evolution domains of TSP have important roles in regulating vascular tissue remodeling: they harbor sequences associated with the anti-angiogenic action of these two proteins[41,42] and with the inhibition of metalloproteinases (MMP)[43,44]. One of the N-terminal domains in TSP-1 contains a sequence that binds TGF-β: TSP-1 is a major activator of latent TGF-β[45-47].
The anti-angiogenic properties of TSP-1 and TSP-2 have been studied for over 30 years[48]. TSP-1 and TSP-2 inhibit endothelial cell proliferation, migration, and apoptosis[49-52]. The decreased expression of TSP-1 and TSP-2 in tumors and surrounding tissues has been reported[53-55], and animal studies confirmed their anti-angiogenic and tumor-preventing action[56]. Furthermore, TSP-1 expression has been associated with cancer dormancy[57,58]; merely suppressing or overexpressing TSP-1 is enough to reverse the patterns of tumor growth in specific anatomical areas with differential expression of TSP-1[59-63].
In addition to the direct effects of TSP-1 and TSP-2 on endothelial cells, the regulation of angiogenesis may be an indirect consequence of regulation of MMP activity[44], binding of growth factors and regulation of their availability and activity[64], and regulation of functions of the immune and inflammatory cells[65,66].
The effects of TSP-1 on cancer cells are sometimes inconsistent with its anti-angiogenic and anti-cancer effects observed in in vivo studies. TSP-1 has adhesive properties that support cancer cell growth[67-69], promote metastatic properties of breast cancer cells[70], facilitate the invasion of squamous cell carcinoma[71], breast cancer cells[72] and melanoma[73], may increase proliferation of cancer cells[74], and decrease cancer cell apoptosis[75].
Integrative approach to understanding TSP roles in cancer
These contradictory effects of TSPs on cultured cancer cells and on the fate of a tumor in vivo have not been explained. To better understand their significance, these contradictory effects should be considered in a context of complex relationships between the cancer cells and the entire organism: prevention of a tumor growth not only relies on the cancer cell properties alone but also requires a concerted response of the body that involves the activation of immune responses, the recognition of cancer cells, and clearance of these cells. Tumor development occurs only when multiple body systems fail to eliminate cancer cells from the system. Cancer cells are constantly forming in different tissues and also circulate in blood[76] but fail to attach and initiate a tumor growth when the microenvironment (including ECM of tissues), regulation of angiogenesis, and responses of the immune system are normal[77,78]. Dysregulation of metabolic, immune, and tissue remodeling processes is what leads a single cancer cell to progress to a tumor rather than being recognized, killed and eliminated. The physiological balance of cancer cell attachment, proliferation and mobility versus their recognition, apoptosis, and elimination define the fate of each cancer cell that forms in the body.
The in vivo effects of TSP-1 and TSP-2, suggest that these proteins activate a whole-organism anti-angiogenic and anti-cancer program that ultimately leads to a decrease in cancer growth or to cancer cell dormancy[58]. Thus, the positive effects of TSP-1 and TSP-2 on cancer cell proliferation could be considered as a part of this program, which initiates anti-cancer defense in multiple body systems. For example, increased proliferation of cancer cells due to TSP-1 signaling may render the cancer cells more susceptible to the elimination by natural killer cells[79]. Furthermore, TSP-1 signaling may facilitate activation of p53, a regulator of apoptosis[80,81]. Promoting cancer cell proliferation and invasion may result in better responses from T-cells due to expression of cancer-specific antigens and their circulation in blood. Similar to the therapeutic approaches designed by humans, e.g., chemotherapy and radiation treatment of cancers, the natural body responses may be the most efficient when the cancer cells are rapidly growing. Understanding why TSPs have cell-specific responses and seemingly contradictory effects would explain how they protect from cancers in the case of TSP-1 or promote cancer growth in the case of TSP-4. Better understanding these complex and sometimes contradictory properties of TSPs will only be possible by developing an integrative approach and more holistic view of the pathological and physiological processes regulated by these proteins, considering the fact that they affect multiple organ systems.
Newly developed integrative approaches to cancer therapies have pushed the field to better understand the causes of cancer and the mechanisms, by which tumors grow and spread. As a result, inflammation and the metabolic changes have become the focus of many studies that investigate how the cancer microenvironment is regulated[82,83]. A growing body of evidence connects increased levels of blood glucose and insulin and chronic inflammation with cancer initiation and progression[84,85]. While the association of diabetes and cancer has been known for many years[86-89], recent studies suggested that even post-prandial elevations in blood glucose and/or insulin increases the risk of cancer. The glycemic load (GL, a measure of the increase in post-prandial blood glucose caused by food) and/or the high dietary glycemic index (GI, another index that estimates the effect of foods on post-prandial blood glucose) were associated with a risk of breast cancer[90-94]; with lung cancer[95]; prostate cancer[93,96], especially with its aggressive form[97]; endometrial cancer[93,98]; ovarian cancer[93]; and digestive tract cancers (esophageal, stomach, colorectal, liver, gallbladder, and pancreatic)[93,96,99-103]. The emerging evidence stresses the importance of diets low in GI and GL and reduction of carbohydrates in diets as a part of healthy nutrition and lifestyle to prevent cancer development and recurrence[104-106]. The connection between chronic inflammation and cancer has been known for a long time: e.g., an association between the hepatitis and the liver cancer has been well recognized and studied[107,108], the existence of cancers caused by pancreatitis and Crohn’s disease has been known and accepted[109,110], and the connection between the infection with italicize and stomach cancer has been confirmed[111,112]. Diabetes, pre-diabetes, and metabolic syndrome are associated with chronic inflammation[113-116] and can be induced by the chronic inflammation in growing adipose tissue[117-119] and pancreas[120-122]. Thus, metabolic dysfunction appears to increase the risk of cancer directly (due to an increased blood glucose and insulin) and by increasing the inflammation. TSP-1, that normally restrains angiogenesis and prevents the growth of a tumor, is downregulated by high blood glucose levels in many tissues[123,124], thus, providing a link between the elevated blood glucose and cancer. TSP-1 has been shown to be downregulated by a microRNA, miR-467, in response to hyperglycemia[124]. Inhibition of miR-467 using an antagonist effectively inhibited hyperglycemia-induced breast cancer growth in mice[125]. Furthermore, decreased levels of TSP-1 are associated with higher inflammation in tissues, probably due to its ability to stimulate phagocytosis in macrophages and to promote the resolution of inflammation[66,126]. Therefore, increasing the levels of TSP-1 may stop or prevent the growth of tumors in multiple complementary ways by decreasing cancer angiogenesis and promoting the resolution of cancer inflammation.
Anti-cancer TSP-based approaches
The functions of TSP-2, TSP-3, and TSP-4 in regulation of cancer growth are not well enough understood to identify potential therapeutic approaches based on the regulation of expression of these proteins or on their specific ligands and cell surface receptors. However, TSP-1, a TSP family member discovered and purified from platelets[127] 40 years ago, has been a target for developing strategies to modulate its levels or to take advantage of its interactions with ligands in ECM and on the cell surface.
Multiple attempts to use TSP-1 fragments to inhibit cancer growth have been described in the literature. Adenovirus-mediated gene therapy containing an antiangiogenic fragment of TSP-1 inhibited the growth of the human leukemia xenograft in mice[128]. Gene therapy with a fragment of TSP-1 inhibited the growth of human breast carcinoma, MDA-MB-435, in vivo in mice[129]. The delivery of the fragment together with p53 resulted in a synergistic effect and decreased the cancer growth more than the TSP-1 fragment or p53 administered separately. Linear and cyclic peptide TSP-1 mimetics have been tested in anti-angiogenesis therapies[130-135].
The interaction of TSP-1 with CD47 was shown to mediate multiple effects of TSP-1[136]. Targeting this interaction, with the goal of increasing angiogenesis, led to an unexpected outcome - angiogenesis inhibition[137]. The peptide, designed to block the interaction of TSP-1 with CD47, named TAX2, increased the binding of TSP-1 to CD36 and disrupted vascular endothelial growth factor receptor 2 activation and subsequent downstream NO signaling. This peptide was also tested in experimental animal cancer models and inhibited angiogenesis and growth of melanoma[137], pancreatic carcinoma[137] and neuroblastoma[138]. It was also effective in preventing the spread of melanoma[139]. The 4N1 peptide, based on the sequence of TSP-1 domain that binds CD47, was successfully used in a mouse model as an anti-leukemia agent[140]. The interaction of TSP-1 with CD47 was found to be important in multiple processes related to tumor growth. For example, blocking the signaling through CD47 conferred protection of normal tissue to irradiation through activation of autophagy pathways[141,142]. Modulation of the anti-tumor immunity by CD47 in T cells by this pathway has been described[143]. Thus, this TSP-1-CD47 interaction appears to be a valuable therapeutic target.
One of the cell-specific effects of TSP-1, mediated by its interaction with CD47, limits cell survival in response to radiation[144], suggesting that antagonizing this interaction would provide a selective radioprotection for normal cells and tissues. Another tissue- and cell-specific approach targeted a miRNA regulating TSP-1 production: miR-467 increases in a cell- and tissue-specific manner in response to hyperglycemia and silences the production of TSP-1[124]. Thus, antagonizing this miRNA slows down the growth of certain cancers without affecting TSP-1 production in response to high glucose in other tissues[125].
Some unexpected outcomes from using anti-TSP-1 strategies highlight the complexity of TSP-1 interactions and its functions. The domains involved in regulating angiogenesis, TGF-β activation, and MMP inhibition are localized in N-terminal part of TSP-1, while interaction with CD47 depends on the C-terminal domain of the protein. However, based on the results of peptide studies, the domains are functionally associated, such that blocking the interaction with one receptor also changes the interactions of distant domains with other receptors and ligands[64,145]. Due to the multiple cell-specific functions, the effects of TSPs on various cells types that are involved in tumor progression should be also taken in the account when considering pharmacological interventions that target the expressions of TSPs or block their interactions with their ligands.
ECM proteins appear to be good targets for therapy because of their extracellular localization and relatively easy availability for drugs. However, very few ECM proteins have become successful therapeutic targets. Most ECM proteins, including TSPs, have a complex multi-domain structure with a number of ligands on the ECM and cell surface. The combined effect of TSP interactions with other ligands and receptors may not only depend on their protein levels in tissues but also on the availability of ligands and receptors on the cell surface. Ultimately, the systemic effects caused by inhibiting TSPs or regulating their production should be considered. Successful strategies need to be based on tissue- and cell-specific evidence such that interactions do not alter the functions or expression of TSPs elsewhere.
Interactions between TSP pathways further complicate the final outcomes. For example, studies of the effects of hyperglycemia on breast cancer suggest that TSP-1-dependent pathways may synergize with TSP-4-dependent pathways. Higher expression of miR-467 in response to high glucose was associated with inhibition of TSP-1 production[123,124,146][Figure 1]. In addition to its anti-angiogenic effects, TSP-1 is a regulator of inflammation and functions of macrophages[147]. TSP-1 is known to regulate the production of cytokines by macrophages[148-150], to stimulate micropinocytosis[151], motility[152], to activate toll-like receptor 4 pathway in macrophages[153], and to promote the resolution of inflammation[150,154]. Hyperglycemia changes the levels of multiple ECM proteins, including the master ECM regulator TGF-β[155,156]. Higher levels of TGF-β have been detected in the cancers of diabetic patients[157,158], and blocking TGF-β signaling leads to better outcomes in animal models[159-161]. It was reported recently that increased levels of TGF-β led to increased production of TSP-4. Unlike TSP-1, TSP-4 is pro-angiogenic[162,163] and increases accumulation of macrophages and other leukocytes in tissues via increased recruitment into tissues[34,35]. Increased levels of TSP-4 combined with decreased levels of TSP-1 would promote a pro-angiogenic and inflammatory microenvironment leading to tumor growth [Figure 1].
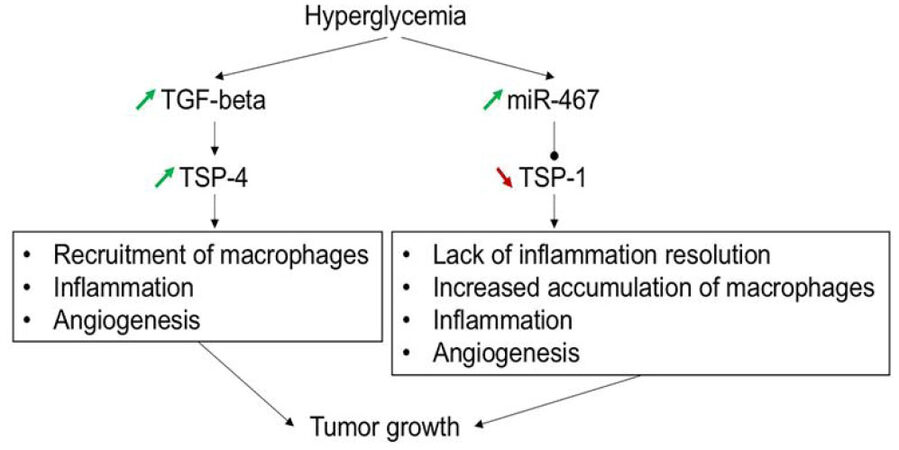
Figure 1. Hyperglycemia promotes cancer growth by regulating thrombospondin (TSP)-1- and TSP-4-dependent pathways. Upregulation of TGF-β in response to hyperglycemia leads to upregulation of TSP-4. TSP-4 is a pro-angiogenic protein that also promotes recruitment of macrophages and other leukocytes into tissues and increases local inflammation. Upregulation of miR-467 in a tissue-specific manner blocks TSP-1 production. In the absence of the anti-angiogenic pressure of TSP-1, cancer angiogenesis is increased. In the absence of TSP-1, the resolution of inflammation is impaired. Increased inflammation and angiogenesis promote cancer growth in the absence of TSP-1. TSP-1 and TSP-4 pathways converge and complement each other to promote the tumor growth
In addition to functional interaction, TSP pathways interact at the mechanistic level. For example, TSP-1 activates TGF-β[45,47,164] and is downregulated by TGF-β[163] but TSP-4 is a mediator of the TGF-β effects[163] and, in turn, modulates the expression of one of the TGF-β receptors, beta-glycan[165], thus, controlling TGF-β signaling [Figure 2].
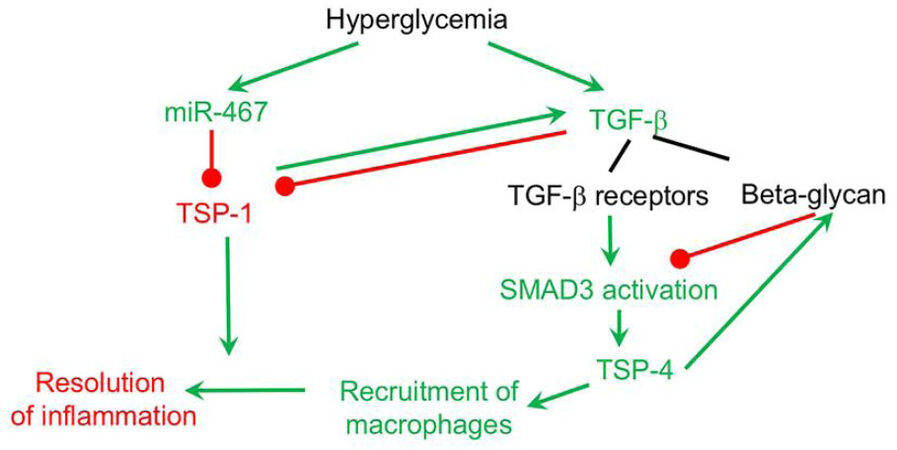
Figure 2. Interaction of hyperglycemia-regulated thrombospondin (TSP) pathways. TSP-4 increases recruitment of macrophages, while TSP-1 is needed for the resolution of inflammation. In response to hyperglycemia, TSP-1 levels are downregulated by increased levels of miR-467. TSP-4 is upregulated as a result of upregulation of TGF-β and activation of SMAD3. Upregulation of TSP-4 result in increased recruitment of macrophages into the tumor. In the absence of TSP-1 and resolution of inflammation, the accumulation of macrophages increases. In a feedback loop, TSP-4 increases the levels of an inhibitory TGF-β receptor beta-glycan. TGF-β further decreases the production of TSP-1. In a feedback loop, TSP-1 is an activator of TGF-β. Green arrow and text = upregulation in response to hyperglycemia; red arrow and text = downregulation in response to hyperglycemia. TGF-β: transforming growth factor beta
Conclusion
TSPs become available to many cell types after they are secreted and incorporated into ECM. They have multiple interactions and functions, which depend on the availability of specific cell surface receptors on each cell type at any given moment. The final outcome of modulating TSP levels is determined by a combined effect from their actions in multiple cell types and organs, from the tumor itself to the immune system and vasculature.
While multiple targets may potentiate the effects of modulation of TSP expression and functions, the complexity of TSP interactions requires an unbiased testing of the effects of potential anti-cancer therapies in in vivo models. When the interactions and mechanisms are dissected and understood, TSPs may become desirable targets for the integrative anti-cancer approaches.
Declarations
Authors’ contributionsAnalyzed the literature, collected the references: Muppala S, Gajeton J, Stenina-Adognravi O
Prepared a draft of the manuscript: Muppala S, Gajeton J
Prepared the final version of the manuscript: Stenina-Adognravi O
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe work on this review has been supported by funds from NIH to Olga Stenina-Adognravi (RO1 HL117216 and RO1 CA177771) and from the American Heart Association to Jasmine Gajeton/Olga Stenina-Adognravi (17PRE33660475).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19:1423-37.
2. Wang M, Zhao J, Zhang L, Wei F, Lian Y, et al. Role of tumor microenvironment in tumorigenesis. J Cancer 2017;8:761-73.
3. Bremnes RM, Al-Shibli K, Donnem T, Sirera R, Al-Saad S, et al. The role of tumor-infiltrating immune cells and chronic inflammation at the tumor site on cancer development, progression, and prognosis: emphasis on non-small cell lung cancer. J Thorac Oncol 2011;6:824-33.
4. Belli C, Trapani D, Viale G, D'Amico P, Duso BA, et al. Targeting the microenvironment in solid tumors. Cancer Treat Rev 2018;65:22-32.
5. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis 2018;9:115.
6. Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer 2015;15:409-25.
7. Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep 2014;15:1243-53.
8. LaValley DJ, Zanotelli MR, Bordeleau F, Wang W, Schwager SC, et al. Matrix stiffness enhances VEGFR-2 internalization, signaling, and proliferation in endothelial cells. Converg Sci Phys Oncol 2017;3:4.
9. Ng CF, Frieboes HB. Model of vascular desmoplastic multispecies tumor growth. J Theor Biol 2017;430:245-82.
10. Neve A, Cantatore FP, Maruotti N, Corrado A, Ribatti D. Extracellular matrix modulates angiogenesis in physiological and pathological conditions. Biomed Res Int 2014; doi: 10.1155/2014/756078.
11. Yang L, Lin PC. Mechanisms that drive inflammatory tumor microenvironment, tumor heterogeneity, and metastatic progression. Semin Cancer Biol 2017;47:185-95.
12. Medrek C, Pontén F, Jirström K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 2012;12:306.
13. Barcenas CH, Raghavendra A, Sinha AK, Syed MP, Hsu L, et al. Outcomes in patients with early-stage breast cancer who underwent a 21-gene expression assay. Cancer 2017;123:2422-31.
14. Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol 2011; doi: 10.1101/cshperspect.a005058.
15. Adams JC, Lawler J. The thrombospondins. Cold Spring Harb Perspect Biol 2011; doi: 10.1101/cshperspect.a009712.
16. Tan K, Lawler J. The interaction of thrombospondins with extracellular matrix proteins. J Cell Commun Signal 2009;3:177-87.
17. Stenina-Adognravi O. Invoking the power of thrombospondins: regulation of thrombospondins expression. Matrix Biol 2014;37:69-82.
18. Hellewell AL, Gong X, Schärich K, Christofidou ED, Adams JC. Modulation of the extracellular matrix patterning of thrombospondins by actin dynamics and thrombospondin oligomer state. Biosci Rep 2015; doi: 10.1042/BSR20140168.
19. Bentley AA, Adams JC. The evolution of thrombospondins and their ligand-binding activities. Mol Biol Evol 2010;27:2187-97.
20. McCart Reed AE, Song S, Kutasovic JR, Reid LE, Valle JM, et al. Thrombospondin-4 expression is activated during the stromal response to invasive breast cancer. Virchows Arch 2013;463:535-45.
21. Posey KL, Hankenson K, Veerisetty AC, Bornstein P, Lawler J, et al. Skeletal abnormalities in mice lacking extracellular matrix proteins, thrombospondin-1, thrombospondin-3, thrombospondin-5, and type IX collagen. Am J Pathol 2008;172:1664-74.
22. Dalla-Torre CA, Yoshimoto M, Lee CH, Joshua AM, de Toledo SR, et al. Effects of THBS3, SPARC and SPP1 expression on biological behavior and survival in patients with osteosarcoma. BMC Cancer 2006;6:237.
23. Cho JY, Lim JY, Cheong JH, Park YY, Yoon SL, et al. Gene expression signature-based prognostic risk score in gastric cancer. Clin Cancer Res 2011;17:1850-7.
24. D'Errico M, de Rinaldis E, Blasi MF, Viti V, Falchetti M, et al. Genome-wide expression profile of sporadic gastric cancers with microsatellite instability. Eur J Cancer 2009;45:461-9.
25. Singh D, Febbo PG, Ross K, Jackson DG, Manola J, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell 2002;1:203-9.
26. Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell 2004;5:607-16.
27. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486:346-52.
28. Turashvili G, Bouchal J, Baumforth K, Wei W, Dziechciarkova M, et al. Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer 2007;7:55.
29. Förster S, Gretschel S, Jöns T, Yashiro M, Kemmner W. THBS4, a novel stromal molecule of diffuse-type gastric adenocarcinomas, identified by transcriptome-wide expression profiling. Mod Pathol 2011;24:1390-403.
30. Su F, Zhao J, Qin S, Wang R, Li Y, et al. Over-expression of thrombospondin 4 correlates with loss of miR-142 and contributes to migration and vascular invasion of advanced hepatocellular carcinoma. Oncotarget 2017;8:23277-88.
31. Myers T, Chengedza S, Lightfoot S, Pan Y, Dedmond D, et al. Flexible heteroarotinoid (Flex-Het) SHetA2 inhibits angiogenesis in vitro and in vivo. Invest New Drugs 2009;27:304-18.
32. Muppala S, Frolova E, Xiao R, Krukovets I, Yoon S, et al. Proangiogenic properties of thrombospondin-4. Arterioscler Thromb Vasc Biol 2015;35:1975-86.
33. Muppala S, Xiao R, Krukovets I, Verbovetsky D, Yendamuri R, et al. Thrombospondin-4 mediates TGF-β-induced angiogenesis. Oncogene 2017;36:5189-98.
34. Pluskota E, Stenina OI, Krukovets I, Szpak D, Topol EJ, et al. Mechanism and effect of thrombospondin-4 polymorphisms on neutrophil function. Blood 2005;106:3970-8.
35. Frolova EG, Pluskota E, Krukovets I, Burke T, Drumm C, et al. Thrombospondin-4 regulates vascular inflammation and atherogenesis. Circ Res 2010;107:1313-25.
36. Frolova EG, Sopko N, Blech L, Popovic ZB, Li J, et al. Thrombospondin-4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB J 2012;26:2363-73.
37. Greco SA, Chia J, Inglis KJ, Cozzi SJ, Ramsnes I, et al. Thrombospondin-4 is a putative tumour-suppressor gene in colorectal cancer that exhibits age-related methylation. BMC Cancer 2010;10:494.
38. van Doorn R, Zoutman WH, Dijkman R, de Menezes RX, Commandeur S, et al. Epigenetic profiling of cutaneous T-cell lymphoma: promoter hypermethylation of multiple tumor suppressor genes including BCL7a, PTPRG, and p73. J Clin Oncol 2005;23:3886-96.
39. Adams JC, Monk R, Taylor AL, Ozbek S, Fascetti N, et al. Characterisation of drosophila thrombospondin defines an early origin of pentameric thrombospondins. J Mol Biol 2003;328:479-94.
41. Iruela-Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD. Inhibition of angiogenesis by thrombospondin-1 is mediated by 2 independent regions within the type 1 repeats. Circulation 1999;100:1423-31.
42. Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med 2002;6:1-12.
43. Bein K, Simons M. Thrombospondin type 1 repeats interact with matrix metalloproteinase 2. Regulation of metalloproteinase activity. J Biol Chem 2000;275:32167-73.
44. Rodriguez-Manzaneque JC, Lane TF, Ortega MA, Hynes RO, Lawler J, et al. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc Natl Acad Sci U S A 2001;98:12485-90.
45. Murphy-Ullrich JE, Schultz-Cherry S, Höök M. Transforming growth factor-beta complexes with thrombospondin. Mol Biol Cell 1992;3:181-8.
46. Schultz-Cherry S, Chen H, Mosher DF, Misenheimer TM, Krutzsch HC, et al. Regulation of transforming growth factor-beta activation by discrete sequences of thrombospondin 1. J Biol Chem 1995;270:7304-10.
47. Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, et al. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 1998;93:1159-70.
48. Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, et al. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci U S A 1990;87:6624-8.
49. Ichii T, Koyama H, Tanaka S, Shioi A, Okuno Y, Otani S, et al. Thrombospondin-1 mediates smooth muscle cell proliferation induced by interaction with human platelets. Arterioscler Thromb Vasc Biol 2002;22:1286-92.
50. Dawson DW, Pearce SF, Zhong R, Silverstein RL, Frazier WA, et al. CD36 mediates the in vitro inhibitory effects of thrombospondin-1 on endothelial cells. J Cell Biol 1997;138:707-17.
51. Guo N, Krutzsch HC, Inman JK, Roberts DD. Thrombospondin 1 and type I repeat peptides of thrombospondin 1 specifically induce apoptosis of endothelial cells. Cancer Res 1997;57:1735-42.
52. Vogel T, Guo NH, Krutzsch HC, Blake DA, Hartman J, et al. Modulation of endothelial cell proliferation, adhesion, and motility by recombinant heparin-binding domain and synthetic peptides from the type I repeats of thrombospondin. J Cell Biochem 1993;53:74-84.
55. Lawler J, Detmar M. Tumor progression: the effects of thrombospondin-1 and -2. Int J Biochem Cell Biol 2004;36:1038-45.
56. Gutierrez LS, Suckow M, Lawler J, Ploplis VA, Castellino FJ. Thrombospondin 1--a regulator of adenoma growth and carcinoma progression in the APC(Min/+) mouse model. Carcinogenesis 2003;24:199-207.
57. Almog N, Henke V, Flores L, Hlatky L, Kung AL, et al. Prolonged dormancy of human liposarcoma is associated with impaired tumor angiogenesis. FASEB J 2006;20:947-9.
58. Naumov GN, Bender E, Zurakowski D, Kang SY, Sampson D, et al. A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst 2006;98:316-25.
59. Giuriato S, Ryeom S, Fan AC, Bachireddy P, Lynch RC, et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc Natl Acad Sci U S A 2006;103:16266-71.
60. Weinstat-Saslow DL, Zabrenetzky VS, VanHoutte K, Frazier WA, Roberts DD, et al. Transfection of thrombospondin 1 complementary DNA into a human breast carcinoma cell line reduces primary tumor growth, metastatic potential, and angiogenesis. Cancer Res 1994;54:6504-11.
61. Streit M, Riccardi L, Velasco P, Brown LF, Hawighorst T, et al. Thrombospondin-2: a potent endogenous inhibitor of tumor growth and angiogenesis. Proc Natl Acad Sci U S A 1999;96:14888-93.
62. Streit M, Velasco P, Brown LF, Skobe M, Richard L, et al. Overexpression of thrombospondin-1 decreases angiogenesis and inhibits the growth of human cutaneous squamous cell carcinomas. Am J Pathol 1999;155:441-52.
63. Sheibani N, Frazier WA. Thrombospondin 1 expression in transformed endothelial cells restores a normal phenotype and suppresses their tumorigenesis. Proc Natl Acad Sci U S A 1995;92:6788-92.
64. Resovi A, Pinessi D, Chiorino G, Taraboletti G. Current understanding of the thrombospondin-1 interactome. Matrix Biol 2014;37:83-91.
65. Lopez-Dee Z, Pidcock K, Gutierrez LS. Thrombospondin-1: multiple paths to inflammation. Mediators Inflamm 2011;2011:296069.
66. Kirsch T, Woywodt A, Klose J, Wyss K, Beese M, et al. Endothelial-derived thrombospondin-1 promotes macrophage recruitment and apoptotic cell clearance. J Cell Mol Med 2010;14:1922-34.
67. Tuszynski GP, Rothman V, Murphy A, Siegler K, Smith L, et al. Thrombospondin promotes cell-substratum adhesion. Science 1987;236:1570-3.
68. Chandrasekaran S, Guo NH, Rodrigues RG, Kaiser J, Roberts DD. Pro-adhesive and chemotactic activities of thrombospondin-1 for breast carcinoma cells are mediated by α3β1 integrin and regulated by insulin-like growth factor-1 and CD98. J Biol Chem 1999;274:11408-16.
69. Albo D, Rothman VL, Roberts DD, Tuszynski GP. Tumour cell thrombospondin-1 regulates tumour cell adhesion and invasion through the urokinase plasminogen activator receptor. Br J Cancer 2000;83:298-306.
70. Yee KO, Connolly CM, Duquette M, Kazerounian S, Washington R, et al. The effect of thrombospondin-1 on breast cancer metastasis. Breast Cancer Res Treat 2009;114:85-96.
71. Pal SK, Nguyen CT, Morita KI, Miki Y, Kayamori K, et al. THBS1 is induced by TGFB1 in the cancer stroma and promotes invasion of oral squamous cell carcinoma. J Oral Pathol Med 2016;45:730-9.
72. Horiguchi H, Yamagata S, Rong Qian Z, Kagawa S, Sakashita N. Thrombospondin-1 is highly expressed in desmoplastic components of invasive ductal carcinoma of the breast and associated with lymph node metastasis. J Med Invest 2013;60:91-6.
73. Borsotti P, Ghilardi C, Ostano P, Silini A, Dossi R, et al. Thrombospondin-1 is part of a slug-independent motility and metastatic program in cutaneous melanoma, in association with VEGFR-1 and FGF-2. Pigment Cell Melanoma Res 2015;28:73-81.
74. Filleur S, Volpert OV, Degeorges A, Voland C, Reiher F, et al. In vivo mechanisms by which tumors producing thrombospondin 1 bypass its inhibitory effects. Genes Dev 2001;15:1373-82.
75. Mirochnik Y, Kwiatek A, Volpert OV. Thrombospondin and apoptosis: molecular mechanisms and use for design of complementation treatments. Curr Drug Targets 2008;9:851-62.
76. Rejniak KA. Circulating tumor cells: when a solid tumor meets a fluid microenvironment. Adv Exp Med Biol 2016;936:93-106.
77. Houghton AN, Guevara-Pati-o JA. Immune recognition of self in immunity against cancer. J Clin Invest 2004;114:468-71.
79. Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. J Exp Med 2012;209:2409-22.
80. Yamauchi M, Imajoh-Ohmi S, Shibuya M. Novel antiangiogenic pathway of thrombospondin-1 mediated by suppression of the cell cycle. Cancer Sci 2007;98:1491-7.
81. Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science 1994;265:1582-4.
82. Wu Y, Zhou BP. Inflammation: a driving force speeds cancer metastasis. Cell Cycle 2009;8:3267-73.
84. Ryu TY, Park J, Scherer PE. Hyperglycemia as a risk factor for cancer progression. Diabetes Metab J 2014;38:330-6.
85. Pisani P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch Physiol Biochem 2008;114:63-70.
86. Joslin EP, Lombard HL, Burrows RE, Manning MD. Diabetes and cancer. N Engl J Med 1959;260:486-8.
87. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, et al. Diabetes and cancer: a consensus report. CA Cancer J Clin 2010;60:207-21.
88. Noto H, Tsujimoto T, Sasazuki T, Noda M. Significantly increased risk of cancer in patients with diabetes mellitus: a systematic review and meta-analysis. Endocr Pract 2011;17:616-28.
89. Barone BB, Yeh HC, Snyder CF, Peairs KS, Stein KB, et al. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: a systematic review and meta-analysis. JAMA 2008;300:2754-64.
90. Larsson SC, Bergkvist L, Wolk A. Glycemic load, glycemic index and breast cancer risk in a prospective cohort of Swedish women. Int J Cancer 2009;125:153-7.
91. Dong JY, Qin LQ. Dietary glycemic index, glycemic load, and risk of breast cancer: meta-analysis of prospective cohort studies. Breast Cancer Res Treat ;126:287-94.
92. Sieri S, Pala V, Brighenti F, Agnoli C, Grioni S. High glycemic diet and breast cancer occurrence in the Italian EPIC cohort. Nutr Metab Cardiovasc Dis 2013;23:628-34.
93. Turati F, Galeone C, Gandini S, Augustin LS, Jenkins DJ, et al. High glycemic index and glycemic load are associated with moderately increased cancer risk. Mol Nutr Food Res 2015;59:1384-94.
94. Mullie P, Koechlin A, Boniol M, Autier P, Boyle P. Relation between breast cancer and high glycemic index or glycemic load: a meta-analysis of prospective cohort studies. Crit Rev Food Sci Nutr 2016;5:152-9.
95. Melkonian SC, Daniel CR, Ye Y, Pierzynski JA, Roth JA, et al. Glycemic index, glycemic load, and lung cancer risk in non-hispanic whites. Cancer Epidemiol Biomarkers Prev 2016;25:532-9.
96. Hu J, La Vecchia C, Augustin LS, Negri E, de Groh M, et al. Glycemic index, glycemic load and cancer risk. Ann Oncol 2013;24:245-51.
97. Hardin J, Cheng I, Witte JS. Impact of consumption of vegetable, fruit, grain, and high glycemic index foods on aggressive prostate cancer risk. Nutr Cancer 2011;63:860-72.
98. Nagle CM, Olsen CM, Ibiebele TI, Spurdle AB, Webb PM, et al. Glycemic index, glycemic load and endometrial cancer risk: results from the Australian national endometrial cancer study and an updated systematic review and meta-analysis. Eur J Nutr 2013;52:705-15.
99. Larsson SC, Giovannucci EL, Wolk A. Prospective study of glycemic load, glycemic index, and carbohydrate intake in relation to risk of biliary tract cancer. Am J Gastroenterol 2016;111:891-6.
100. Sieri S, Krogh V, Agnoli C, Ricceri F, Palli D, et al. Dietary glycemic index and glycemic load and risk of colorectal cancer: results from the EPIC-Italy study. Int J Cancer 2015;136:2923-31.
101. Sieri S, Agnoli C, Pala V, Grioni S, Brighenti F, et al. Dietary glycemic index, glycemic load, and cancer risk: results from the EPIC-Italy study. Sci Rep 2017;7:9757.
102. Abe H, Aida Y, Ishiguro H, Yoshizawa K, Miyazaki T, et al. Alcohol, postprandial plasma glucose, and prognosis of hepatocellular carcinoma. World J Gastroenterol 2013;19:78-85.
103. Keum N, Yuan C, Nishihara R, Zoltick E, Hamada T, et al. Dietary glycemic and insulin scores and colorectal cancer survival by tumor molecular biomarkers. Int J Cancer 2017;140:2648-56.
104. Dehghan M, Mente A, Zhang X, Swaminathan S, Li W, et al. Associations of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): a prospective cohort study. Lancet 2017;390:2050-62.
105. Augustin LS, Kendall CW, Jenkins DJ, Willett WC, Astrup A, et al. Glycemic index, glycemic load and glycemic response: an international scientific consensus summit from the international carbohydrate quality consortium (ICQC). Nutr Metab Cardiovasc Dis 2015;25:795-815.
106. Augustin LS, Libra M, Crispo A, Grimaldi M, De Laurentiis M, et al. Low glycemic index diet, exercise and vitamin D to reduce breast cancer recurrence (DEDiCa): design of a clinical trial. BMC Cancer 2017;17:69.
107. Takano S, Yokosuka O, Imazeki F, Tagawa M, Omata M. Incidence of hepatocellular carcinoma in chronic hepatitis B and C: a prospective study of 251 patients. Hepatology 1995;21:650-5.
108. De Vos Irvine H, Goldberg D, Hole DJ, McMenamin J. Trends in primary liver cancer. Lancet 1998;351:215-6.
109. Kirkegård J, Mortensen FV, Cronin-Fenton D. Chronic pancreatitis and pancreatic cancer risk: a systematic review and meta-analysis. Am J Gastroenterol 2017;112:1366-72.
110. Weedon DD, Shorter RG, Ilstrup DM, Huizenga KA, Taylor WF. Crohn's disease and cancer. N Engl J Med 1973;289:1099-103.
111. Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, et al. Overexpression of interleukin-1β induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008;14:408-19.
114. Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol 2016;12:15-28.
115. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 2011;11:98-107.
116. Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol 2011;11:738-49.
117. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 1995;95:2409-15.
118. Choe SS, Huh JY, Hwang IJ, Kim JI, Kim JB. Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front Endocrinol (Lausanne) 2016;7:30.
119. Rutkowski JM, Stern JH, Scherer PE. The cell biology of fat expansion. J Cell Biol 2015;208:501-12.
120. Montane J, Cadavez L, Novials A. Stress and the inflammatory process: a major cause of pancreatic cell death in type 2 diabetes. Diabetes Metab Syndr Obes 2014;7:25-34.
121. Wang W, Guo Y, Liao Z, Zou DW, Jin ZD, et al. Occurrence of and risk factors for diabetes mellitus in Chinese patients with chronic pancreatitis. Pancreas 2011;40:206-12. in Chinese
122. Ewald N, Kaufmann C, Raspe A, Kloer HU, Bretzel RG, et al. Prevalence of diabetes mellitus secondary to pancreatic diseases (type 3c). Diabetes Metab Res Rev 2012;28:338-42.
123. Bhattacharyya S, Marinic TE, Krukovets I, Hoppe G, Stenina OI. Cell type-specific post-transcriptional regulation of production of the potent antiangiogenic and proatherogenic protein thrombospondin-1 by high glucose. J Biol Chem 2008;283:5699-707.
124. Bhattacharyya S, Sul K, Krukovets I, Nestor C, Li J, et al. Novel tissue-specific mechanism of regulation of angiogenesis and cancer growth in response to hyperglycemia. J Am Heart Assoc 2012;1:e005967.
125. Krukovets I, Legerski M, Sul P, Stenina-Adognravi O. Inhibition of hyperglycemia-induced angiogenesis and breast cancer tumor growth by systemic injection of microRNA-467 antagonist. FASEB J 2015;29:3726-36.
126. Jiménez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, et al. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med 2000;6:41-8.
127. Lawler JW, Slayter HS, Coligan JE. Isolation and characterization of a high molecular weight glycoprotein from human blood platelets. J Biol Chem 1978;253:8609-16.
128. Liu P, Wang Y, Li YH, Yang C, Zhou YL, et al. Adenovirus-mediated gene therapy with an antiangiogenic fragment of thrombospondin-1 inhibits human leukemia xenograft growth in nude mice. Leuk Res 2003;27:701-8.
129. Xu M, Kumar D, Stass SA, Mixson AJ. Gene therapy with p53 and a fragment of thrombospondin I inhibits human breast cancer in vivo. Mol Genet Metab 1998;63:103-9.
130. Chan LY, Craik DJ, Daly NL. Cyclic thrombospondin-1 mimetics: grafting of a thrombospondin sequence into circular disulfide-rich frameworks to inhibit endothelial cell migration. Biosci Rep 2015; doi: 10.1042/BSR20150210.
131. Yap R, Veliceasa D, Emmenegger U, Kerbel RS, McKay LM, et al. Metronomic low-dose chemotherapy boosts CD95-dependent antiangiogenic effect of the thrombospondin peptide ABT-510: a complementation antiangiogenic strategy. Clin Cancer Res 2005;11:6678-85.
132. Henkin J, Volpert OV. Therapies using anti-angiogenic peptide mimetics of thrombospondin-1. Expert Opin Ther Targets 2011;15:1369-86.
133. Haviv F, Bradley MF, Kalvin DM, Schneider AJ, Davidson DJ, et al. Thrombospondin-1 mimetic peptide inhibitors of angiogenesis and tumor growth: design, synthesis, and optimization of pharmacokinetics and biological activities. J Med Chem 2005;48:2838-46.
134. Huang H, Campbell SC, Bedford DF, Nelius T, Veliceasa D, et al. Peroxisome proliferator-activated receptor gamma ligands improve the antitumor efficacy of thrombospondin peptide ABT510. Mol Cancer Res 2004;2:541-50.
135. Punekar S, Zak S, Kalter VG, Dobransky L, Punekar I, et al. Thrombospondin 1 and its mimetic peptide ABT-510 decrease angiogenesis and inflammation in a murine model of inflammatory bowel disease. Pathobiology 2008;75:9-21.
136. Rogers NM, Sharifi-Sanjani M, Csányi G, Pagano PJ, Isenberg JS. Thrombospondin-1 and CD47 regulation of cardiac, pulmonary and vascular responses in health and disease. Matrix Biol 2014;37:92-101.
137. Jeanne A, Sick E, Devy J, Floquet N, Belloy N, et al. Identification of TAX2 peptide as a new unpredicted anti-cancer agent. Oncotarget 2015;6:17981-8000.
138. Jeanne A, Martiny L, Dedieu S. Thrombospondin-targeting TAX2 peptide impairs tumor growth in preclinical mouse models of childhood neuroblastoma. Pediatr Res 2017;81:480-8.
139. Jeanne A, Boulagnon-Rombi C, Devy J, Théret L, Fichel C, et al. Matricellular TSP-1 as a target of interest for impeding melanoma spreading: towards a therapeutic use for TAX2 peptide. Clin Exp Metastasis 2016;33:637-49.
140. Li G, Wu H, Cui L, Gao Y, Chen L, et al. CD47-retargeted oncolytic adenovirus armed with melanoma differentiation-associated gene-7/interleukin-24 suppresses in vivo leukemia cell growth. Oncotarget 2015;6:43496-507.
141. Maxhimer JB, Soto-Pantoja DR, Ridnour LA, Shih HB, Degraff WG, et al. Radioprotection in normal tissue and delayed tumor growth by blockade of CD47 signaling. Sci Transl Med 2009; doi: 10.1126/scitranslmed.3000139.
142. Soto-Pantoja DR, Miller TW, Pendrak ML, DeGraff WG, Sullivan C, et al. CD47 deficiency confers cell and tissue radioprotection by activation of autophagy. Autophagy 2012;8:1628-42.
143. Soto-Pantoja DR, Terabe M, Ghosh A, Ridnour LA, DeGraff WG, et al. CD47 in the tumor microenvironment limits cooperation between antitumor T-cell immunity and radiotherapy. Cancer Res 2014;74:6771-83.
144. Isenberg JS, Maxhimer JB, Hyodo F, Pendrak ML, Ridnour LA, et al. Thrombospondin-1 and CD47 limit cell and tissue survival of radiation injury. Am J Pathol 2008;173:1100-12.
145. Jeanne A, Schneider C, Martiny L, Dedieu S. Original insights on thrombospondin-1-related antireceptor strategies in cancer. Front Pharmacol 2015;6:252.
146. Krukovets I, Legerski M, Sul P, Stenina-Adognravi O. Inhibition of hyperglycemia-induced angiogenesis and breast cancer tumor growth by systemic injection of microRNA-467 antagonist. FASEB J 2015;29:3726-36.
147. Moura R, Tjwa M, Vandervoort P, Van Kerckhoven S, Holvoet P, et al. Thrombospondin-1 deficiency accelerates atherosclerotic plaque maturation in ApoE-/- mice. Circ Res 2008;103:1181-9.
148. Xing T, Wang Y, Ding WJ, Li YL, Hu XD, et al. Thrombospondin-1 production regulates the inflammatory cytokine secretion in THP-1 cells through NF-κB signaling pathway. Inflammation 2017;40:1606-21.
149. Stein EV, Miller TW, Ivins-O'Keefe K, Kaur S, Roberts DD. Secreted thrombospondin-1 regulates macrophage interleukin-1β production and activation through CD47. Sci Rep 2016;6:19684.
150. Zhao Y, Xion, Z, Lechner EJ, Klenotic PA, Hamburg BJ, et al. Thrombospondin-1 triggers macrophage IL-10 production and promotes resolution of experimental lung injury. Mucosal Immunol 2013;7:440-8.
151. Csányi G, Feck DM, Ghoshal P, Singla B, Lin H, et al. CD47 and Nox1 mediate dynamic fluid-phase macropinocytosis of native LDL. Antioxid Redox Signal 2017;26:886-901.
152. Liu Z, Morgan S, Ren J, Wang Q, Annis DS, et al. Thrombospondin-1 (TSP1) contributes to the development of vascular inflammation by regulating monocytic cell motility in mouse models of abdominal aortic aneurysm. Circ Res 2015;117:129-41.
153. Li Y, Qi X, Tong X, Wang S. Thrombospondin 1 activates the macrophage toll-like receptor 4 pathway. Cell Mol Immunol 2013;10:506-12.
154. Peter MR, Jerkic M, Sotov V, Douda DN, Ardelean DS, et al. Impaired resolution of inflammation in the endoglin heterozygous mouse model of chronic colitis. Mediators Inflamm 2014; doi: 10.1155/2014/767185.
155. Wu L, Derynck R. Essential role of TGF-beta signaling in glucose-induced cell hypertrophy. Dev Cell 2009;17:35-48.
156. Wheeler SE, Lee NY. Emerging roles of transforming growth factor β signaling in diabetic retinopathy. J Cell Physiol 2017;232:486-9.
157. Melzer C, Hass R, von der Ohe J, Lehnert H, Ungefroren H. The role of TGF-β and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun Signal 2017;15:19.
158. Wintrob ZA, Hammel JP, Nimako GK, Gaile DP, Forrest A, et al. Dataset on growth factor levels and insulin use in patients with diabetes mellitus and incident breast cancer. Data Brief 2017;11:183-91.
159. Park SY, Kim MJ, Park SA, Kim JS, Min KN, et al. Combinatorial TGF-β attenuation with paclitaxel inhibits the epithelial-to-mesenchymal transition and breast cancer stem-like cells. Oncotarget 2015;6:37526-43.
160. Kim D, Lee AS, Jung YJ, Yang KH, Lee S, et al. Tamoxifen ameliorates renal tubulointerstitial fibrosis by modulation of estrogen receptor α-mediated transforming growth factor-β1/Smad signaling pathway. Nephrol Dial Transplant 2014;29:2043-53.
161. Park CY, Min KN, Son JY, Park SY, Nam JS, et al. An novel inhibitor of TGF-β type I receptor, IN-1130, blocks breast cancer lung metastasis through inhibition of epithelial-mesenchymal transition. Cancer Lett 2014;351:72-80.
162. Muppala S, Frolova E, Xiao R, Krukovets I, Yoon S, et al. Proangiogenic properties of thrombospondin-4. Arterioscler Thromb Vasc Biol 2015;35:1975-86.
163. Muppala S, Xiao R, Krukovets I, Verbovetsky D, Yendamuri R, et al. Thrombospondin-4 mediates TGF-β-induced angiogenesis. Oncogene 2017;36:5189-98.
164. Murphy-Ullrich JE, Poczatek M. Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev 2000;11:59-69.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Stenina-Adognravi O, Muppala S, Gajeton J. Thrombospondins and remodeling of the tumor microenvironment. Vessel Plus 2018;2:30. http://dx.doi.org/10.20517/2574-1209.2018.40
AMA Style
Stenina-Adognravi O, Muppala S, Gajeton J. Thrombospondins and remodeling of the tumor microenvironment. Vessel Plus. 2018; 2: 30. http://dx.doi.org/10.20517/2574-1209.2018.40
Chicago/Turabian Style
Stenina-Adognravi, Olga, Santoshi Muppala, Jasmine Gajeton. 2018. "Thrombospondins and remodeling of the tumor microenvironment" Vessel Plus. 2: 30. http://dx.doi.org/10.20517/2574-1209.2018.40
ACS Style
Stenina-Adognravi, O.; Muppala S.; Gajeton J. Thrombospondins and remodeling of the tumor microenvironment. Vessel Plus. 2018, 2, 30. http://dx.doi.org/10.20517/2574-1209.2018.40
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 19
likes
Like This Article 19
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.