
Histone deacetylases in vascular permeability and remodeling associated with acute lung injury

Abstract
Acute lung injury (ALI) is a severe progressive disorder that arises from a wide range of causes such as toxins or inflammation, resulting in significant morbidity and mortality. There are no effective therapeutic options apart from mechanical ventilation strategies. While the mechanisms that govern the clinically relevant process of increased endothelial cell (EC) permeability and remodeling associated with ALI are under intense investigation, our knowledge of the processes that determine barrier enhancement or preservation are far from completion. Recently, epigenetic mechanisms have emerged as a major regulator of enduring changes in cell behavior and the therapeutic potential of inhibiting histone deacetylases (HDACs) for the treatment of cardiovascular and inflammatory diseases has gained remarkable attention. Although HDACs have been shown to play an important role in regulating EC barrier function, the involved HDAC subtypes and mechanisms remain undefined. Further investigation of the HDAC signaling may provide therapeutic approaches for the prevention and treatment of ALI.
Keywords
Introduction
Acute lung injury (ALI) is a significant source of morbidity and mortality with over 200,000 incidences per year in the US[1,2]. ALI arises from a wide range of causes such as toxins or inflammation resulting in significant morbidity and frequently in death[3]. To date no pharmacological therapies have been proven to improve ALI in clinical trials[4]. Approaches remain non-specific and rely on supportive care and control of initial causes[5]. A major pathophysiological alteration of ALI is the dysfunction of the pulmonary vascular endothelial barrier resulting in pulmonary infiltrates, hypoxemia and pulmonary edema[6,7]. Endothelial cells (ECs) line the lumen of the blood vessels serving every organ system and provide a semi-selective barrier between the blood and the interstitial space[8]. In ALI, the EC barrier is compromised leading to elevated vascular permeability[9]. A lack of understanding the pathologic mechanisms involved in ALI remains an obstacle to new and effective therapies that reduces the vascular leakage in ALI.
Recently, epigenetic mechanisms have gained great therapeutic potential in the treatment of inflammatory and autoimmune diseases[10,11]. Previous studies have demonstrated that inflammatory gene expression is regulated by the precise balance between histone-modulating enzymes; histone acetyltransferases (HATs) and histone deacetylases (HDACs)[12,13]. In addition to modulating histone protein, it has been shown that HDACs can also deacetylate many non-histone proteins to regulate cellular functions such as cytoskeletal acetylation and polymerization, and signal transduction[14-16]. Acetylation of non-histone proteins might crosstalk with other posttranslational modifications (PTMs) to control cellular signaling[17]. HDACs are classes of enzymes that catalyze the removal of the acetyl groups from specific lysine residues of the histone or non-histone proteins[18]. The inhibition of HDACs has been shown to play an important role in EC barrier protection[19,20]. In this review, we intend to summarize the current knowledge focusing on the potential effect of HDAC inhibition on the pathogenesis of the ALI.
Characterization of HDACs
There are 18 characterized members of HDAC superfamily in human which can be divided into two families and four classes according to their function and homology to yeast proteins[21][Table 1]. The classical family includes the classes I, II, and IV HDACs which are zinc-dependent enzymes and perform metal ion-mediated hydrolysis of acetamide bond in their acetylated substrates while silent information regulator 2 (Sir2)-related protein (Sirtuin) family contains the class III HDACs requiring NAD+ as a cofactor for enzymatic activity[22,23]. Mechanisms for HDAC regulation occurs at multiple levels involving protein complex formation, PTMs such as phosphorylation, SUMOylation, subcellular localization, availability of metabolic cofactors and proteolytic processing[24].
Characterization of HDACs
| Class | HDAC isoform | Size (amino acids) | Subcellular localization | Expression pattern |
|---|---|---|---|---|
| I | HDAC1 | 482 | Nucleus | Ubiquitous |
| HDAC2 | 488 | Nucleus | Ubiquitous | |
| HDAC3 | 428 | Mainly nucleus | Ubiquitous | |
| HDAC8 | 377 | Nucleus/cytoplasm | Ubiquitous | |
| IIa | HDAC4 | 1084 | Nucleus/cytoplasm | Heart, skeletal muscle, brain |
| HDAC5 | 1122 | Nucleus/cytoplasm | Heart, skeletal muscle, brain | |
| HDAC7 | 952 | Nucleus/cytoplasm | Heart, placenta, pancreas, skeletal muscle | |
| HDAC9 | 1011 | Nucleus/cytoplasm | Skeletal muscle, brain | |
| IIb | HDAC6 | 1215 | Mainly cytoplasm | Heart, liver, kidney, pancreas |
| HDAC10 | 669 | Mainly cytoplasm | Liver, spleen, kidney | |
| III | SIRT1 | 747 | Nucleus | Ubiquitous |
| SIRT2 | 352 | Cytoplasm | Ubiquitous | |
| SIRT3 | 399 | Mitochondria | Ubiquitous | |
| SIRT4 | 314 | Mitochondria | Ubiquitous | |
| SIRT5 | 310 | Mitochondria | Ubiquitous | |
| SIRT6 | 355 | Nucleus | Ubiquitous | |
| SIRT7 | 400 | Nucleolus | Ubiquitous | |
| IV | HDAC11 | 347 | Mainly nucleus | Brain, heart, skeletal muscle, kidney and testis |
The class I HDACs (HDAC1, 2, 3, and 8) have sequence similarity to the yeast transcriptional regulator reduced potassium dependency 3 (RPD3) protein. They are ubiquitously expressed and mainly localized within the cellular nucleus[25]. HDAC1 and HDAC2 contain nuclear localization signal (NLS), but not nuclear export signal (NES), therefore they are restricted to the nucleus[26]. HDAC3 are able to shuttle between the nucleus and the cytoplasm which is regulated by competing nuclear import and nuclear export signals[27]. HDAC8 shows relatively low expression and can be found in the nucleus and in the cytoplasm[28]. Class I HDACs are ~350-500 amino acids long. They consist of the conserved deacetylase domain (DAC) with short amino- and carboxy-terminal tails, latter can be modified by PTMs including phosphorylation or SUMOylation in order to regulate the enzymatic activity[24]. HDACs 1-3 form multiprotein complexes with transcription factors and co-repressors to fulfil their deacetylase activities. HDAC8 does not form complexes and is fully active in single molecule[29,30].
The class II HDACs are protein orthologous to the yeast histone deacetylase-A 1 (HDA1) protein and can be divided into two subclasses such as class IIa (HDAC4, 5, 7 and 9) and class IIb (HDAC6 and 10)[25]. Class II HDACs show tissue-specific expression pattern and functions[31]. They are able to shuttle between nucleus and cytoplasm, however, HDAC6 and 10 are predominantly localized in the cytoplasm[32]. Class II HDACs are considerably larger molecules than class I HDACs, and they have approximately 700-1200 amino acid residues. The class IIa HDACs have the deacetylase domain on the C-terminal and possess a long N-terminal tail containing conserved multiple binding domains and regulatory sites which play a crucial role in the regulation of the nucleocytoplasmic trafficking[29,33]. The catalytic domain of class IIb HDACs is localized on the N-terminus of the protein. In addition, the HDAC6 has a secondary DAC domain and an ubiquitin binding site on the C-terminal[34]. HDAC10 is closely related to HDAC6 and has a putative second catalytic domain and two putative Rb binding domains on the C-terminal of the enzyme[35].
The HDAC11 shares sequence similarity to both RPD3 and HDA1 proteins and falls into the Class IV as a unique member. It is the smallest HDAC isoform consisting 347 amino acid residues and are located predominantly in the nucleus. HDAC11 has tissue-specific distribution and can be found in the brain, heart, skeletal muscle kidney and testis. It contains a catalytic domain at the N-terminus and short N- and C-terminal extensions[36].
The class III HDACs have seven members (SIRT1, 2, 3, 4, 5, 6 and 7) and possess homology to the yeast Sir2 protein[37]. They are ubiquitously expressed and show different subcellular localization. SIRT1, 6 and 7 are mainly localized to the nucleus, the SIRT2 can be found in cytoplasm, while the SIRT3, 4 and 5 are mitochondrial proteins[38]. They do not comprise zinc in the catalytic site and uses NAD+ as a cofactor in their catalytic reactions. The 275 amino acid long catalytic domain is highly conserved among the sirtuins flanking with variable length of amino- and carboxy-terminal extensions[39]. They can serve as a NAD+-dependent lysine deacetylase or as a mono-ADP-ribosyltransferase. In the deacetylation reaction, nicotinamide, 2’-O-acetyl-ADP-ribose and deacetylated product are generated with the hydrolysis of one NAD+ molecule[40]. During the ADP-ribosylation reaction, ADP-ribose from the NAD+ is transferred to the acetylated substrate and nicotinamide is released[41]. Both reactions depend on the ratio of NAD+/NADH. Therefore the cellular metabolism can be a potential regulatory mechanism of SIRTs.
Biological function of HDACs
Lysine acetylation/deacetylation of histone and non-histone proteins are major reversible PTMs that are dynamically maintained by two enzymes families, HAT and HDAC[42]. Changes in histone acetylation modify chromatin structure and the activity of transcription factors that serve as an important mechanism for the regulation of gene expression[43]. Deacetylation of histones by HDACs contributes to the compaction of the chromatin structure correlating with gene silencing[44]. Genome-wide mapping revealed that HDACs are also associated with active genes and positively correlated with gene transcription, indicating that they do not only remove the acetyl group in active genes but also reset the chromatin for activation of gene transcription[45]. HDACs deacetylate transcriptional factors mostly repressing the gene expression. However, HDACs can also activate transcription by either reducing the transcription of transcriptional repressor proteins or by deacetylating and activating the transcription activators or by deacetylating and inhibiting the transcription repressors[21]. In addition to regulation of gene expression, activities of HDACs have been shown to indirectly regulate other PTMs including phosphorylation, ubiquitination, methylation, SUMOylation, NEDDilation, biotinylation, etc.[17]. Lysine acetylation can interfere with other lysine modifications, conversely, removal of the acetyl group can promote other lysine modifications that control vital cellular functions such as mRNA integrity, translation, enzymatic activity as well as protein stability, function, localization and interactions[46]. For example, there is a direct competition between acetylation and ubiquitination, namely acetylation blocks ubiquitination and proteasome-mediated degradation of target proteins. Similarly, the deacetylation facilitates protein degradation[47]. Members of the HDAC superfamily have been shown to be involved in many physiological processes such as cell migration, proliferation and survival, cell differentiation, cell cycle, signal transduction, aging, DNA repair and apoptosis[48,49]. Growing body of evidence show that malfunction of the HDACs play a critical role in many human disorders including cancer, neurodegenerative diseases, metabolic and immunological disorders, inflammatory, cardiac and pulmonary diseases[50,51]. HDAC inhibition has been reported to display antitumor and anti-inflammation properties[52-55]. It has also been shown that HDACs play an important role in various inflammatory lung diseases including ALI, COPD, and asthma[56-58].
HDACs in ALI
Several reports have indicated that HDAC inhibitors possess beneficial effect in ALI animal models. The primary causes of ALI are the endothelial barrier dysfunction and inflammation[59,60]. It has been shown that HDAC6-specific inhibitor tubacin ameliorates pulmonary edema in the LPS-induced ALI[19]. Pharmacological inhibition of HDAC6 suppresses the thrombin-induced endothelial barrier dysfunction through increased acetylation of α-tubulin and microtubules stabilization[19]. In addition, Yu et al.[61] found that selective inhibition of HDAC6 by tubastatin A (Tub A) blocks TNF-α-induced lung endothelial permeability and prevents endotoxin-induced pulmonary edema. Pretreatment with Tub A enhanced α-tubulin acetylation and decreased the TNF-α-induced microtubule disassembly, endothelial cell contraction and actin stress fiber formation as well as reduced the phosphorylation of the myosin light chain attenuating the lung endothelial cell hyperpermeability caused by the cytokine[61]. In addition, HDAC6 inhibition by Tub A increases β-catenin acetylation and consequently its membrane translocation leading to increased stabilization of adherens junctions in endothelial cells[61]. Moreover, inhibition of HDAC6 prevents endotoxin-induced deacetylation of α-tubulin and β-catenin in lung tissues and attenuates lung edema formation in mouse model of endotoxemia[61]. The same group demonstrated that selective inhibitors of HDAC6 such as CAY10603 and Tub A prevented the TNF-α-induced caspase 3 activation and endothelial barrier dysfunction via maintaining cell-cell junction integrity[62]. In addition, inhibition of HDAC6 by CAY10603 alleviated the endotoxin-induced lung vascular permeability and caspase-3 activation as well as reduced the lung edema formation[62]. Joshi et al.[20] revealed the mediating role of HDACs in LPS-induced endothelial hyperpermeability and ALI. Inhibition of various HDACs by pan-HDAC inhibitors including panobinostat or trichostatin (TSA) attenuates LPS-induced decrease in transendothelial electrical resistance (TER), Hsp90 activation and chaperone function as wells as diminishes the RhoA activity and signaling. Moreover, pre-treatment with HDAC3-selective inhibitor RGFP-966 or with HDAC6- selective inhibitor Tub A, or combined inhibition of HDAC3 and -6 prevents the LPS-mediated endothelial barrier dysfunction[20]. More importantly, combined pharmacological inhibition of HDAC3 and -6 protected against LPS-stimulated inflammation, capillary permeability, and structural abnormalities in murine model of ALI[20].
HDAC inhibition has been shown to ameliorate the inflammatory responses associated with ALI. Ni et al.[63] reported that broad HDAC inhibitor, butyrate markedly diminishes the pulmonary inflammation in LPS-induced ALI in mice. LPS administration induces histopathological changes in murine lungs, increases the production of TNF-α, IL-1β and NO, as well as increases MPO activity and NF-κB p65 expression that are significantly attenuated by butyrate pretreatment[63]. The lung edema is markedly reduced by butyrate, indicating its protective effect on the LPS-induced ALI[63]. Zhang et al.[64] further confirmed the beneficial effect of butyrate on lung injury. They found that two structurally unrelated HDAC inhibitors, sodium butyrate (SB) and TSA diminished the sepsis-induced lung edema and leukocyte infiltration in lung tissue[64]. In addition, SB and TSA decrease the expression of ICAM-1 and E-selectin in lung tissue and reduces plasma levels of IL-6, indicating that these HDAC inhibitors attenuate sepsis-induced inflammatory lung injury[64]. Other group has also demonstrated that early administration of broad-spectrum HDAC inhibitor valproic acid (VPA) significantly reduces the levels of IL-6 and tumor necrosis factor in bronchoalveolar lavage (BAL) fluid and in plasma as well as improves survival in murine ALI model in gram-negative pneumonia[65]. They also showed that VPA reduces the production of proinflammatory cytokines and the neutrophil influx into the pulmonary parenchyma and alleviates the host systemic and pulmonary inflammatory responses[66]. In addition, the HDAC inhibitor VPA decreases the MPO activity reducing the lung injury induced by the bacterial infection and improves the histopathologic changes related to ALI[66]. VPA has also been reported to prevent ALI induced by ischemia-reperfusion (I/R) in rat lungs. I/R significantly increases the lung edema, pulmonary arterial pressure, lung inflammation and the concentrations of the inflammatory mediators [TNF-α, cytokine-induced neutrophil chemoattractant-1 (CINC-1)] in bronchoalveolar lavage fluid (BALF). Pretreatment with VPA diminishes the I/R-caused alterations via increased heme oxygenase-1 (HO-1) activity that is an essential protective regulator in lung injury[67]. Lu et al.[68] demonstrated that HDAC inhibitors, suberanilohydroxamic acid (SAHA) and its analogue 4-(dimethylamino)-N-[7-(hydroxyamino)-7-oxoheptyl] benzamide significantly decreases early neutrophilic inflammation in murine model of ALI. They showed that these inhibitors are able to block the leukotriene A4 hydrolase (LTA4H) activity and to prevent the leukotriene B4 (LTB4) biosynthesis suppressing the LPS-induced neutrophils migration and infiltration into the murine lungs as well as reduces the production of inflammatory mediator (e.g., TNF-α, IL-1β, and IL-6) induced by LPS[68].
HDAC6 plays an important role in the cigarette smoke extract (CSE)-induced lung endothelial barrier disruption[69,70]. Cigarette smoke (CS) activates HDAC6 that results in the deacetylation of α-tubulin and microtubule destabilization, leading to the impairment of lung endothelial barrier function and the exacerbation of LPS- or P. aeruginosa-induced elevation in lung vascular endothelial permeability[70]. Down-regulation of HDAC6 by tubacin or by siRNAs to HDAC6 significantly reduces the CSE-induced elevation in the endothelial permeability in vitro. In addition, inhibition of HDAC6 attenuates the lung inflammation and lung edema induced by CSE in LPS- or P. aeruginosa-induced ALI animal model, indicating the involvement of HDAC6 in the CS exacerbation of LPS- or P. aeruginosa-induced ALI[70].
Conclusion
In summary, convincing evidence support that members of the HDAC family play a critical role in the development of ALI [Figure 1]. Manipulating the HDAC signaling pathways using multidisciplinary approaches would provide novel therapeutic strategies for the protection of endothelial barrier function and for the intervention of ALI in inflammatory lung diseases.
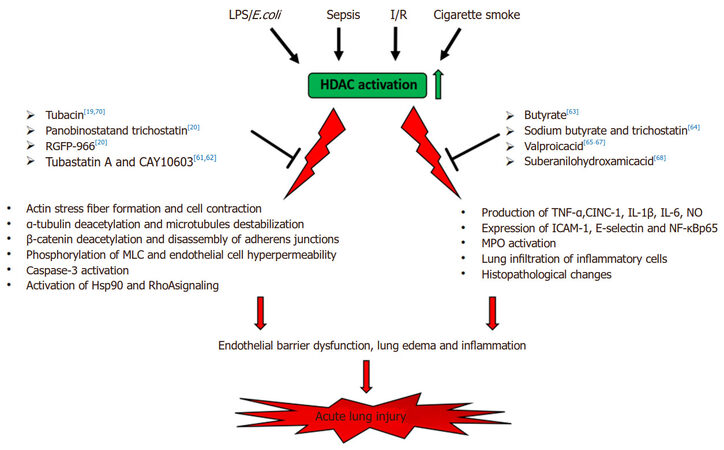
Figure 1. Schematic illustration of role of HDACs in acute lung injury (ALI). HDAC inhibitors: Tubacin (N1-[4-[(2R,4R,6S)-4-[[(4,5-diphenyl-2-oxazolyl)thio]methyl]-6-[4-(hydroxymethyl)phenyl]-1,3-dioxan-2-yl]phenyl]-N8-hydroxy-octanediamide)[19,70]; Panobinostat (NVP-LBH589)[20]; Trichostatin A (TSA, 7-[4-(dimethylamino)phenyl]-N-hydroxy-4,6R-dimethyl-7-oxo-2E,4E-heptadienamide)[20,64]; RGFP-966 ((2E)-N-(2-Amino-4-fluorophenyl)-3-[(2E)-1-(3-phenyl-2-propen-1-yl)-1H-pyrazol-4-yl]-2-propenamide)[20]; Tubastatin A (N-hydroxy-4-[(1,2,3,4-tetrahydro-2-methyl-5H-pyrido[4,3-b]indol-5-yl)methyl]-benzamide)[61,62]; CAY10603 (N-[4-[3-[[[7-(hydroxyamino)-7-oxoheptyl]amino]carbonyl]-5-isoxazolyl]phenyl]-1,1-dimethylethyl ester, carbamic acid)[61,62]; Sodium Butyrate (butanoic acid sodium salt)[63,64]; Valproic acid (VPA, 2-Propylpentanoic acid)[65-67]; Vorinostat (Suberoylanilide Hydroxamic Acid, SAHA)[68]. LPS: lipopolysaccharides; E.coli: Escherichia coli; I/R: ischemia-reperfusion; MLC: myosin light chain; TNF-α: tumor necrosis factor-α; CINC-1: cytokine-induced neutrophil chemoattractant-1; IL-1β: interleukin 1β; IL-6: interleukin 6; NO: nitric oxide; ICAM-1: intercellular adhesion molecule 1; NF-κB p65: nuclear factor kappa-B p65 subunit; MPO: myeloperoxidase
Declarations
Authors’ contributionsConcept/design: Kovacs L, Kovacs-Kasa A, Su Y
Draft: Kovacs L, Kovacs-Kasa A
Manuscript editing and review: all authors
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by NIH/NHLBI R01 HL134934 (YS), VA Merit Review Award BX002035 (YS), Flight Attendants Medical Research Institute grant 140083_CIA (YS), AHA Career Development Award 18CDA34110225 (LK) and AHA Postdoctoral Fellowship 18POST33990193 (AKK).
Conflicts of interestAll authors declare that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv 2010;23:243-52.
2. Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 2005;353:1685-93.
3. Butt Y, Kurdowska A, Allen TC. Acute lung injury: a clinical and molecular review. Arch Pathol Lab Med 2016;140:345-50.
5. Gonzales JN, Lucas R, Verin AD. The acute respiratory distress syndrome: mechanisms and perspective therapeutic approaches. Austin J Vasc Med 2015;2:1009.
6. Maniatis NA, Kotanidou A, Catravas JD, Orfanos SE. Endothelial pathomechanisms in acute lung injury. Vascul Pharmacol 2008;49:119-33.
7. Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol 2009;77:1763-72.
8. Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 2004;84:869-901.
9. Kasa A, Csortos C, Verin AD. Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers 2015;3:e974448.
10. Jeffries MA, Sawalha AH. Autoimmune disease in the epigenetic era: how has epigenetics changed our understanding of disease and how can we expect the field to evolve? Expert Rev Clin Immunol 2015;11:45-58.
11. Greer JM, McCombe PA. The role of epigenetic mechanisms and processes in autoimmune disorders. Biologics 2012;6:307-27.
12. Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev 1998;12:599-606.
13. Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 1998;20:615-26.
15. Halili MA, Andrews MR, Sweet MJ, Fairlie DP. Histone deacetylase inhibitors in inflammatory disease. Curr Top Med Chem 2009;9:309-19.
16. Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 2000;20:6891-903.
17. Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell 2008;31:449-61.
19. Saito S, Lasky JA, Guo W, Nguyen H, Mai A, Danchuk S, Sullivan DE, Shan B. Pharmacological inhibition of HDAC6 attenuates endothelial barrier dysfunction induced by thrombin. Biochem Biophys Res Commun 2011;408:630-4.
20. Joshi AD, Barabutis N, Birmpas C, Dimitropoulou C, Thangjam G, Cherian-Shaw M, Dennison J, Catravas JD. Histone deacetylase inhibitors prevent pulmonary endothelial hyperpermeability and acute lung injury by regulating heat shock protein 90 function. Am J Physiol Lung Cell Mol Physiol 2015;309:L1410-9.
21. Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 2014;6:a018713.
22. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003;370:737-49.
23. Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. Sirtuins: the 'magnificent seven', function, metabolism and longevity. Ann Med 2007;39:335-45.
25. Bjerling P, Silverstein RA, Thon G, Caudy A, Grewal S, Ekwall K. Functional divergence between histone deacetylases in fission yeast by distinct cellular localization and in vivo specificity. Mol Cell Biol 2002;22:2170-81.
26. Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov 2002;1:287-99.
27. Yang WM, Tsai SC, Wen YD, Fejer G, Seto E. Functional domains of histone deacetylase-3. J Biol Chem 2002;277:9447-54.
28. Van den Wyngaert I, de Vries W, Kremer A, Neefs J, Verhasselt P, Luyten WH, Kass SU. Cloning and characterization of human histone deacetylase 8. FEBS Lett 2000;478:77-83.
29. Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 2008;9:206-18.
30. Watson PJ, Millard CJ, Riley AM, Robertson NS, Wright LC, Godage HY, Cowley SM, Jamieson AG, Potter BV, Schwabe JW. Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat Commun 2016;7:11262.
31. Verdin E, Dequiedt F, Kasler HG. Class II histone deacetylases: versatile regulators. Trends Genet 2003;19:286-93.
32. Bertos NR, Wang AH, Yang XJ. Class II histone deacetylases: structure, function, and regulation. Biochem Cell Biol 2001;79:243-52.
33. Parra M, Verdin E. Regulatory signal transduction pathways for class IIa histone deacetylases. Curr Opin Pharmacol 2010;10:454-60.
34. Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol 2008;18:291-7.
35. Guardiola AR, Yao TP. Molecular cloning and characterization of a novel histone deacetylase HDAC10. J Biol Chem 2002;277:3350-6.
36. Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem 2002;277:25748-55.
37. Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun 2000;273:793-8.
38. Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev 2006;20:2913-21.
39. Min J, Landry J, Sternglanz R, Xu RM. Crystal structure of a SIR2 homolog-NAD complex. Cell 2001;105:269-79.
41. Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun 1999;260:273-9.
42. Drazic A, Myklebust LM, Ree R, Arnesen T. The world of protein acetylation. Biochim Biophys Acta 2016;1864:1372-401.
44. Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol 2007;8:983-94.
45. Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009;138:1019-31.
46. Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 2009;41:185-98.
47. Caron C, Boyault C, Khochbin S. Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. Bioessays 2005;27:408-15.
48. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 2009;10:32-42.
49. Bassett SA, Barnett MP. The role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients 2014;6:4273-301.
50. Lawless MW, Norris S, O'Byrne KJ, Gray SG. Targeting histone deacetylases for the treatment of disease. J Cell Mol Med 2009;13:826-52.
51. Tang J, Yan H, Zhuang S. Histone deacetylases as targets for treatment of multiple diseases. Clin Sci (Lond) 2013;124:651-62.
52. Pan LN, Lu J, Huang B. HDAC inhibitors: a potential new category of anti-tumor agents. Cell Mol Immunol 2007;4:337-43.
53. Walkinshaw DR, Yang XJ. Histone deacetylase inhibitors as novel anticancer therapeutics. Curr Oncol 2008;15:237-43.
55. Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, Dona G, Fossati G, Sozzani S, Azam T, Bufler P, Fantuzzi G, Goncharov I, Kim SH, Pomerantz BJ, Reznikov LL, Siegmund B, Dinarello CA, Mascagni P. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci U S A 2002;99:2995-3000.
56. Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, Barnes PJ. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med 2005;352:1967-76.
57. Ito K, Charron CE, Adcock IM. Impact of protein acetylation in inflammatory lung diseases. Pharmacol Ther 2007;116:249-65.
58. Adcock IM, Ito K, Barnes PJ. Histone deacetylation: an important mechanism in inflammatory lung diseases. COPD 2005;2:445-55.
59. Chong DL, Sriskandan S. Pro-inflammatory mechanisms in sepsis. Contrib Microbiol 2011;17:86-107.
60. Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003;101:3765-77.
61. Yu J, Ma Z, Shetty S, Ma M, Fu J. Selective HDAC6 inhibition prevents TNF-alpha-induced lung endothelial cell barrier disruption and endotoxin-induced pulmonary edema. Am J Physiol Lung Cell Mol Physiol 2016;311:L39-47.
62. Yu J, Ma M, Ma Z, Fu J. HDAC6 inhibition prevents TNF-alpha-induced caspase 3 activation in lung endothelial cell and maintains cell-cell junctions. Oncotarget 2016;7:54714-22.
63. Ni YF, Wang J, Yan XL, Tian F, Zhao JB, Wang YJ, Jiang T. Histone deacetylase inhibitor, butyrate, attenuates lipopolysaccharide-induced acute lung injury in mice. Respir Res 2010;11:33.
64. Zhang L, Jin S, Wang C, Jiang R, Wan J. Histone deacetylase inhibitors attenuate acute lung injury during cecal ligation and puncture-induced polymicrobial sepsis. World J Surg 2010;34:1676-83.
65. Kasotakis G, Galvan MD, Osathanugrah P, Dharia N, Bufe L, Breed Z, Mizgerd JP, Remick DG. Timing of valproic acid in acute lung injury: prevention is the best therapy? J Surg Res 2017;220:206-12.
66. Kasotakis G, Galvan M, King E, Sarkar B, Stucchi A, Mizgerd JP, Burke PA, Remick D. Valproic acid mitigates the inflammatory response and prevents acute respiratory distress syndrome in a murine model of Escherichia coli pneumonia at the expense of bacterial clearance. J Trauma Acute Care Surg 2017;82:758-65.
67. Wu SY, Tang SE, Ko FC, Wu GC, Huang KL, Chu SJ. Valproic acid attenuates acute lung injury induced by ischemia-reperfusion in rats. Anesthesiology 2015;122:1327-37.
68. Lu W, Yao X, Ouyang P, Dong N, Wu D, Jiang X, Wu Z, Zhang C, Xu Z, Tang Y, Zou S, Liu M, Li J, Zeng M, Lin P, Cheng F, Huang J. Drug repurposing of histone deacetylase inhibitors that alleviate neutrophilic inflammation in acute lung injury and idiopathic pulmonary fibrosis via inhibiting leukotriene A4 hydrolase and blocking LTB4 biosynthesis. J Med Chem 2017;60:1817-28.
69. Lu Q, Sakhatskyy P, Grinnell K, Newton J, Ortiz M, Wang Y, Sanchez-Esteban J, Harrington EO, Rounds S. Cigarette smoke causes lung vascular barrier dysfunction via oxidative stress-mediated inhibition of RhoA and focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol 2011;301:L847-57.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Kovacs L, Kovacs-Kasa A, Verin AD, Fulton D, Lucas R, Su Y. Histone deacetylases in vascular permeability and remodeling associated with acute lung injury. Vessel Plus 2018;2:15. http://dx.doi.org/10.20517/2574-1209.2018.06
AMA Style
Kovacs L, Kovacs-Kasa A, Verin AD, Fulton D, Lucas R, Su Y. Histone deacetylases in vascular permeability and remodeling associated with acute lung injury. Vessel Plus. 2018; 2: 15. http://dx.doi.org/10.20517/2574-1209.2018.06
Chicago/Turabian Style
Kovacs, Laszlo, Anita Kovacs-Kasa, Alexander D. Verin, David Fulton, Rudolf Lucas, Yunchao Su. 2018. "Histone deacetylases in vascular permeability and remodeling associated with acute lung injury" Vessel Plus. 2: 15. http://dx.doi.org/10.20517/2574-1209.2018.06
ACS Style
Kovacs, L.; Kovacs-Kasa A.; Verin AD.; Fulton D.; Lucas R.; Su Y. Histone deacetylases in vascular permeability and remodeling associated with acute lung injury. Vessel Plus. 2018, 2, 15. http://dx.doi.org/10.20517/2574-1209.2018.06
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 3 clicks
Cite This Article 3 clicks

 Like This Article 15
likes
Like This Article 15
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.