
Energetic metabolism in cardiomyocytes: molecular basis of heart ischemia and arrhythmogenesis


Abstract
Cardiac muscle contraction is a strictly regulated process which conjugates a series of electrophysiological, biochemical and mechanic events, resulting in the pumping of blood to all bodily tissues. These phenomena require a very high energetic demand both for generating the necessary mechanical force, and for maintaining cellular homeostasis during the process. In the myocardium, fatty acids (FA) represent the main energy substrate, although other secondary substrates, such as glucose and ketone bodies, may also be used. Nevertheless, under certain conditions such as heart failure or myocardial ischemia, FA metabolism may become deleterious via mechanisms such as oxidative stress and arrhythmogenesis. In an ischemic milieu, various metabolic changes occur as a consequence of hypoxia, favoring cell necrosis, ventricular arrhythmias, and death. Major events in this context include an increase in extracellular K+, a decrease in pH, and accumulation of intracellular calcium. This review includes a detailed description of the molecular basis underlying myocardial contraction and energetic metabolism in cardiomyocytes, aiming to promote an integral understanding of the pathophysiology of heart ischemia. This in turn may aid in the development of future, more satisfactory alternative treatments in the management of acute coronary ischemia episodes.
Keywords
Introduction
The heart is composed of specialized muscle tissue, the myocardium, which depends on a constant input of energy for adequate functioning, and thus utilizes various sources of carbon for adenosine triphosphate (ATP) production. However, in cardiomyocytes, the majority of the energy produced derives from the mitochondrial oxidation of free fatty acids (FA)[1].
Alterations in myocardial energetic metabolism may lead to conditions such as ischemic heart disease, arrhythmias, heart failure, and others. During ischemia, cardiomyocytes are restricted in both their oxygen and nutrient supplies, rendering these cells unable to oxidize energetic substrates, causing a significant depletion in ATP reserves. This leads to deterioration of cardiac contractility and disruptions in the cardiac conduction system, which may culminate in heart failure and arrhythmias, respectively[2]. Therefore, an understanding of myocardial energetic metabolism is essential, in order to comprehend the physiologic basis of the therapeutic management of acute and chronic heart disease.
Fuel for myocardial contraction: the role of macromolecules
The heart is the driving force of the circulatory system, pumping blood to all bodily tissues. This organ consists of various layers, the thickest of which is the myocardium. The main component of this layer is contractile cells termed cardiomyocytes[3], with only 2% corresponding to Purkinje fibers. The Purkinje fibers are organized into an arborized structure which originates in the atrioventricular node and constitutes a specialized conduction system that allows quick and synchronic activation of the ventricles[4,5].
The cytosol of cardiomyocytes contains sarcomeres, complex proteic structures composed of thick myosin filaments and thin actin filaments[6]. Sarcomeres occupy most of these cells’ cytosol and mediate calcium-dependent cellular contraction [Figure 1][7]. This phenomenon is regulated by the cardiac conduction system through modulation of ionic transport and generation of action potentials[8]. These impulses are synchronically propagated throughout the myocardium via gap junctions and ionic channels which facilitate electrical and chemical communication between cardiomyocytes, allowing these to function as a syncytium[9]. Connexins, especially Cx43, Cx40 and Cx45 are key voltage-gated proteic structures found in these gap junctions, each of which can differentially and dynamically intervene in the propagation of the action potential[10-12].
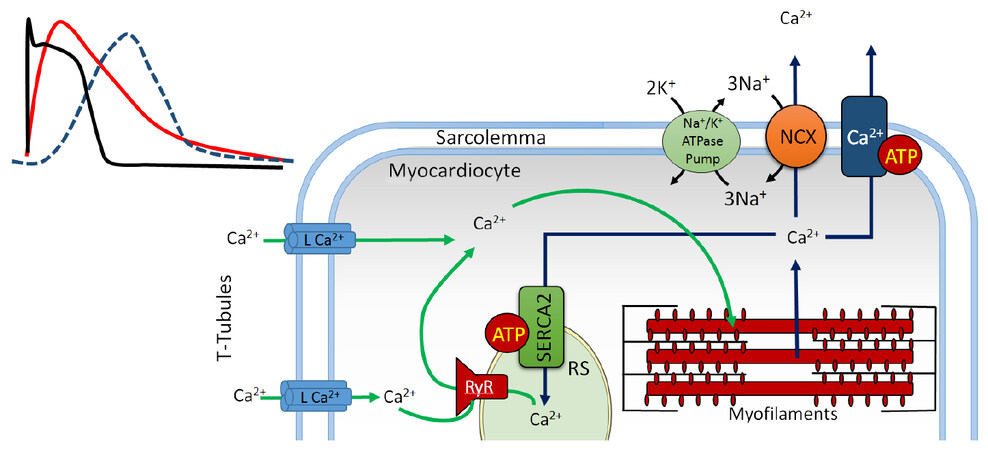
Figure 1. The role of calcium in excitation-contraction coupling. Green arrows: calcium entry; blue arrows: calcium exit; SR: sarcoplasmic reticulum; RyR: ryanodine receptor; NCX: Na+/Ca2+ exchanger
The myocardium is a form of specialized striated muscle, richly innervated by the autonomic nervous system, which is under uninterrupted activity throughout life. Therefore, it requires a constant and substantial energetic input from macromolecules such as carbohydrates, lipids and proteins in order to sustain the process of contraction and relaxation. Indeed, the cardiomyocyte transforms chemical energy from FA, glucose, ketone bodies and other substrates into mechanical energy[13,14]. The energetic metabolism of the cardiomyocyte consists of three key components: (1) capture and utilization of primary substrates, with the incorporation of their metabolites into the tricarboxylic acid (TCA) cycle; (2) oxidative phosphorylation, which occurs in the respiratory chain within the internal mitochondrial membrane; and (3) the phosphocreatine-creatine kinase energy transference system, a network for phosphate transference from ATP to creatine (an "energy-storing" molecule), through mitochondrial creatine kinase and yielding phosphocreatine, an important source of energy under high-demand conditions[15].
The metabolic machinery of the heart utilizes oxygen up to 80%-90% of the maximum capacity of the electron transport chain; however, at a resting state, the heart operates at only 15%-25% of its maximum oxidative capacity[16]. Cardiomyocytes show an elevated rate of ATP hydrolysis, which is strongly linked to oxidative phosphorylation. Because under non-ischemic conditions, over 95% of these cells’ ATP is produced in this process[17], it is indispensable in order to assure the full replenishment of the cardiomyocytes’ ATP content every 10 s, and thus maintain constant concentrations of this molecule, even under conditions of increased frequency or force of contractions[18]. Of the total energy produced by ATP hydrolysis, approximately 60%-70% serves as fuel for contraction, while the remaining 30%-40% is used by the Ca2+ATPase pumps in the smooth sarcoplasmic reticulum and other ion pumps[19].
FA metabolism in cardiomyocytes
Cardiomyocytes use FA as their main source of energy, yet their synthesizing capacity for these molecules is relatively low[20] as is shown in Figure 2. As a result, these cells depend fundamentally on the influx of FA from the vascular compartment, and thus, the rate of FA consumption by cardiac muscle is principally determined by the concentration of non-esterified FA in plasma[21,22].
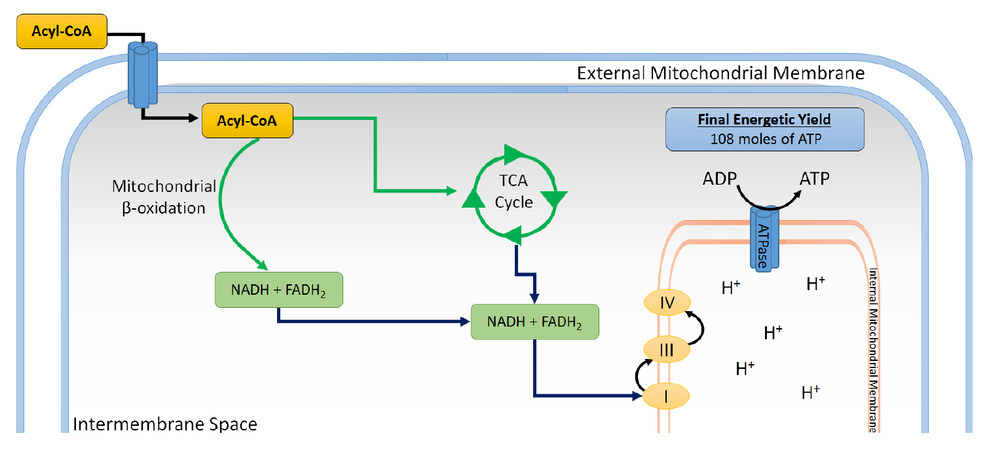
Figure 2. FA metabolism in myocardiocytes. Once inside cells, FA are esterified with Coenzyme A into acyl-CoA, which in turn can be esterified for storage as triacylglycerides, or transported to the external mitochondrial membrane, where it is carried by a carnitine-dependent system into the intermembrane space. In this process, the acyl group is condensed with carnitine and separated from CoA to form acyl-carnitine, via carnitine palmitoyl transferase I. Then, it binds to acyl-carnitine translocase, which transports it into the mitochondrial matrix, and is converted into acetyl CoA, which goes into the TCA cycle. This process also yields NADH and FADH2 and several reducing equivalents, which can be utilized in the respiratory chain to generate ATP through oxidative phosphorylation. FA: fatty acid; TCA: tricarboxylic acid
The heart obtains these FA chiefly from exogenous substrates, especially free FA bound to albumin, and FA released from triacylglycerides (TAG) contained in chylomicrons and very low-density lipoproteins, later undergoing β-oxidation[23-25]. FA may enter cardiomyocytes by passive diffusion or by active transport through the sarcolemma, involving an FA translocase (CD36) or an FA-binding protein in the cell membrane[26]. This translocation is mediated by intracellular vesicles, and may be promoted by electrical stimulation or high-demand conditions[27].
CD36 is one of the main translocases, an 80 kD integral membrane glycoprotein which is stored in intracellular compartments and transported towards the cell membrane in response to increased energy demands[28]. This protein is found in platelets, immune cells, adipocytes, myocytes, enterocytes and various other cells, yet is most abundantly expressed in cardiomyocytes. It is also the most important FA translocase in the heart and plays a key role in the entry of long-chain FA into cardiomyocytes[29]. The extracellular domain of CD36 has three disulfide bridges in its C-terminus end which contain binding sites for FA, oxidized low-density lipoprotein, thrombospondin and Plasmodium falciparum-infected erythrocytes. This domain operates actively by generating transduction signals which interact with multiple tyrosine-kinases and generate various structural modifications which modulate the functions of CD36; including phosphorylation, glycosylation, palmitoylation and ubiquitination[30].
As cardiac activity increases, so does the cardiomyocyte’s metabolic demand, which results in an increase in intracellular concentrations of adenosine monophosphate (AMP) and reactive oxygen species[31]. In turn, this upregulates AMP-activated protein kinase (AMPK), a key metabolic regulator. AMPK is a serine/threonine kinase which acts as a metabolic sensor in cardiomyocytes. It is activated during high energy requirement states, enhancing FA availability, uptake and oxidation in these cells by promoting the expression and activation of lipoprotein lipase and CD36, also known as fatty acid translocase[31]. This favors the entry of long-chain FA into the cell, preserving stable levels of ATP in the face of increased metabolic demand[32]. Nevertheless, excessive expression of CD36 has been associated with impaired cardiac insulin sensitivity, reduced uptake of glucose, and excessive uptake of FA, subsequently causing cardiomyocyte lipotoxicity and retention of GLUT4 in their cytoplasm[33]. Recent in vitro studies in cardiomyocytes have shown that use of CD36 blockers or deletion of its coding gene ameliorates contractile dysfunction mediated by lipotoxicity, and reduced lipid-induced damage[34,35]. AMPK can also inhibit acetyl-CoA carboxylase, which enhances mitochondrial FA uptake. In addition, in energy depletion states, AMPK increases GLUT4 expression and inhibits its internalization and also enhances glycolysis by phosphorylation of phosphofructokinase 2. It may also facilitate glycogen storage in adequate ATP supply states[36].
In addition to their plasma concentration, an important long-term regulator of FA β-oxidation is the modulation by peroxisome proliferator-activated receptor (PPAR)[37]. Numerous coactivator proteins, such as PPAR-γ co-activator 1-α can powerfully induce the transcription of PPAR target genes, including those involved in FA storage (such as diacylglycerol acyltransferase, promoted by PPARα), FA oxidation (such as medium-chain acyl-CoA dehydrogenase, promoted by PPARα/β/δ/γ), and glucose metabolism (such as pyruvate dehydrogenase kinase 4, promoted by PPARα)[38,39].
PPAR also plays an important role in the regulation of oxidative stress in the cardiovascular system, with several isoforms implicated in various transcriptional mechanisms for antioxidant genes[40,41]. For example, PPARα and PPARγ promote the transcription and activation of Cu/Zn-superoxide dismutase (SOD1), Mn-superoxide dismutase (SOD2) and catalase in cardiac tissue. Furthermore, PPARα augments IGF-1 transcription, subsequently activating the IGF-1/PI3K pathway, inhibiting apoptosis and protecting cardiomyocytes under ischemic stress[42].
Carbohydrate metabolism in cardiomyocytes
Carbohydrates are also a valuable source of energy in the myocardium, with glucose providing roughly a quarter of the total energy produced in a well-irrigated heart. Of this total ATP, approximately 10%-40% derives from the oxidation of glucose-lactate within the TCA, and only 2% derives from glycolysis[43].
Glucose enters the cardiomyocyte through glucose transporter proteins (GLUT). Fourteen different GLUTs have been described in humans, all of which appear to be able to transport hexoses or polyols, although it is suspected that many other GLUT substrates remain undiscovered[44]. GLUT 1-5 are the most studied to date, and they are well-known to be glucose and/or fructose transporters in various tissues and cell types[45]. In cardiomyocytes, GLUT4 is the main transporter, translocating to the membrane in response to signaling by insulin, increased work demand, or ischemia, with GLUT1 playing an accessory role[14].
The products of glycolysis are utilized in both the TCA cycle and the respiratory chain in order to generate ATP through oxidative phosphorylation[46]. Although only 2% the heart’s ATP is produced in glycolysis, it becomes very important under anaerobic or ischemic conditions. Indeed, in heart failure and hypertrophy, there is a metabolic switch towards favoring carbohydrate over FA metabolism in the heart, with a notable change being the acceleration of glycolysis[47]. This increase in the glycolytic flux appears to be due to a functional upregulation in the pathway’s enzyme, rather than a clear increase in the expression of glycolytic enzymes[48].
This shift towards utilization of glucose in the hypertrophic myocardium had traditionally been considered a maladaptive change. Nevertheless, recent studies in bioengineering-modified mice have demonstrated glucose-dependence not to be harmful in adult hearts, and a decrease in the utilization of glucose appears to be deleterious in failure and hypertrophy[49]. For example, mice with GLUT1 overexpression - and thus, increased glycolysis - appear to be protected against heart failure and left ventricular dilatation, even when subjected to pressure overload[50,51]. On the other hand, those with deletion of GLUT4 and insulin receptors in the heart failure, and showed worse responses to cardiac hypertrophy-promoting stimuli[52].
The phosphocreatine-creatine kinase system incardiomyocytes
Because both the systole and diastole are active, highly ATP-dependent processes, the demand for these metabolites in cardiomyocytes is very high, with the heart requiring approximately 20 times its weight in ATP per day in order to sustain its energetic demands[53]. After synthesis, ATP must be transported from the mitochondria to the myofilaments and membrane proton pumps, a process in which the phosphocreatine-creatine kinase system intervenes substantially[54].
Creatine kinase (CK) is a key enzyme for phosphate transference in cells with high energetic demand, and works in harmony with other enzymatic machinery in order to facilitate intracellular energetic communication[55]. CK synthesizes phosphocreatine (PC) from creatine and a phosphate group from ATP in a reversible reaction, acting as a functional ATP reserve. CK associated with myofilaments catalyzes the transference of the phosphate from PC towards adenosine diphosphate (ADP), replenishing ATP in ATPase active sites, such as myosin heads. In cardiomyocytes, CK isoenzymes and the highly diffusible PC are responsible for sustaining the transference of energy from producing centers (mitochondria and glycolysis) towards ATP-consuming sites (myofilaments and ATPase pumps)[15].
The PC-CK system represents the first line of energetic reserves in cardiomyocytes, providing a quick source of ATP and favoring its transportation to its utilization sites, especially myofilaments[56]. In animal models, disruptions in the PC-CK system have been linked to impaired myocardial contractility and increased risk for arrhythmias[57,58]. Moreover, alterations in the functionality of CK have been identified as an independent risk factor for heart failure[59].
CK is composed of dimers, which consist of subunits M and B, and originate three isoenzymes: CK-MM, -MB and -BB. A fourth isoenzyme is found in mitochondria (mi-CK) and accounts for 20%-40% of all CK activity in the heart[60]. CK is not evenly distributed within the cell, and is rather a part of a compartmentalized metabolic pathway, bound to myofilaments and the sarcoplasmic reticulum to form functional complexes which accelerate ATP synthesis[61].
The mi-CK isoform is coupled to the external surface of the internal mitochondrial membrane, near the ATP-ADP translocases, also termed adenine nucleotide translocases (ANT). During oxidative phosphorylation, the ATP generated in the mitochondrial matrix is exported by ANT to the intermembrane space and transphophorylated by mi-CK to PC and ADP, with the latter being immediately available for oxidative phosphorylation, stimulating cellular respiration[55]. Another isoform of CK is associated with myofilaments, acting as a structural protein within M-bands, which is functionally coupled to the myosin ATPase and can transfer phosphate from PC to ATP, providing sufficient energy for maintaining maximum contractility[61].
In a healthy heart, approximately two thirds of all creatine is phosphorylated by CK to yield PC. In heart failure, the level of PC is lower in relation to the concentrations of ATP, with a lower PC/ATP index[62]. Lower values of this index have been related to increased mortality[63]. Human and animal models have demonstrated a progressive reduction in the creatine pool of up to 60% in patients with heart failure, with a directly proportional relationship between the decrease of the index and the severity of the condition[64].
Fatty acids vs. glucose as energetic substrates
The selection of energetic substrates in cardiomyocytes is a fundamental step for the constant generation of ATP which depends on the dynamic metabolic milieu in each body at a given time[65]. This flexibility is present during fetal development; however, after birth, FA becomes the preferential substrates, due to the increased availability of oxygen and dietary fats[66-68]. Infants with mutations in genes involved in FA metabolism have been documented to develop cardiomyopathy when under stress, highlighting the essential role of FA in this tissue[66]. Likewise, in heart failure and left ventricular hypertrophy (when the oxidative capacity of mitochondria in cardiomyocytes is diminished), there is a shift towards a predominance for glucose metabolism[69,70].
FA are known to be the main source of energy in cardiomyocytes when the heart is at rest and during fasting periods: most of the acetyl CoA that enters the TCA cycle (60%-90%) originates in the β-oxidation of free FA[13], while the remaining 10%-40% is produced by oxidation of pyruvate, which derives from the oxidation of glucose or lactic acid[71]. Several reports have shown that cardiac efficiency, in terms of oxygen consumption, is greater when oxidizing glucose and lactate rather than FA[72]. Studies using ranolazine - an inhibitor of β-oxidation which induces oxidation of carbohydrates - have found enhanced left ventricular function and improved metabolic efficiency when utilizing glucose as the main energetic substrate[73]. Similarly, potentiating FA use in the heart with heparin or TAG infusions has been reported to result in a 26% increase in oxygen consumption without changes in mechanical capacity of the left ventricle[74], which suggests a greater functional capacity for this chamber when utilizing glucose[75]. This may be due to the higher level of oxidative stress caused by the oxidation of FA in comparison with carbohydrates, due to the increased oxygen consumption rate in the former[76].
The ATP synthesis/oxygen consumption rates for glucose and lactic acid are 3.17 and 3.00, respectively; whereas they are 2.80 and 2.86 for palmitate and oleate, respectively. Although these are theoretical values which may be lower in vivo as a consequence of the constant proton efflux through the mitochondrial membrane, the differences between substrates remain substantial[77]. For example, when comparing palmitate with glucose, the complete oxidation of 1 molecule of palmitate yields 105 ATP molecules and requires 46 oxygen atoms, while 1 molecule of glucose generates 31 ATP molecules and uses 12 oxygen atoms. Thus, despite FA clearly yielding greater amount of ATP, this occurs at the expense of larger oxygen requirements[39]. Furthermore, β-oxidation of FA generates more lipid peroxidation due to increased delivery of NADH and FADH2 to the electron transport chain and production of superoxide anion[78].
In addition, elevated free FA are harmful in the ischemic myocardium, augmenting cell damage. In the first hours of an acute myocardial infarction (AMI), free FA can act as detergents on the cell membrane of cardiomyocytes[79-81]. Moreover, there is increased generation of free radicals, which can inactivate IRS-1 via phosphorylation of serine residues. This directly promotes insulin resistance and simultaneously stimulates the release of proinflammatory cytokines, such as TNF-α and IL-6[82]. Therefore, all conditions of metabolic inefficiency in the heart favor insulin-mediated left ventricular remodeling and diastolic myocardial dysfunction[83].
Considering this, various systemic conditions such as obesity cause elevated serum free FA which can potentiate β-oxidation, and thus increase lipid traffic in cardiomyocytes, prompting a phenomenon termed lipotoxicity[13,84]. This process can lead the cell towards contractile dysfunction, insulin resistance and ultimately apoptosis in association with accumulation of ceramides[85].
On the other hand, pre-clinical and clinical evidence suggests partial inhibition of free FA oxidation in the myocardium can prevent or diminish tissue damage and dysfunction under conditions of ischemia or reperfusion, diabetic cardiomyopathy, and AMI. This occurs because the heart shifts towards glucose as the main source for ATP synthesis, which reduces the oxygen demand by 11%-13% and therefore improves cardiac efficiency and protects mitochondrial function[43,86]. Nonetheless, the real benefits of this partial inhibition remain uncertain when contemplating the potential consequences of excessive lipid accumulation within cardiomyocytes[86].
Arrhythmogenic metabolic changes during myocardial ischemia
Cardiac sudden death is responsible for approximately half of all cardiovascular mortality[87]. The majority of these are attributed to ventricular tachyarrhythmias (ventricular tachycardia, ventricular fibrillation), which are frequently caused by myocardial ischemia[88]. The mechanisms through which myocardial ischemia leads to local electrophysiological disorders and arrhythmogenesis have been extensively explored [Figure 3][2,89].
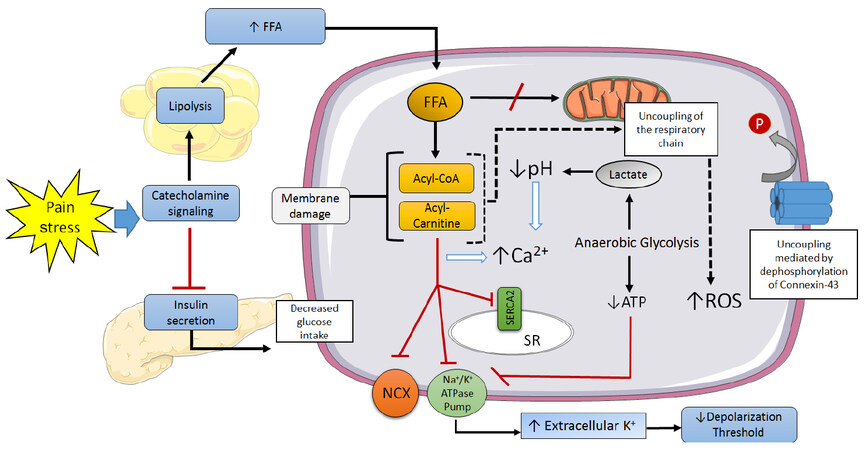
Figure 3. Mechanisms involved in the genesis of ventricular arrhythmias in the ischemic myocardiocyte. SR: sarcoplasmic reticulum; NCX: Na+/Ca2+ exchanger; ROS: reactive oxygen species; FFA: free fatty acids; P: phosphate
Severe metabolic changes begin a few seconds after coronary occlusion: high-energy phosphates are hydrolyzed, intracellular pH lowers as a consequence of the activation of anaerobic glycolysis, and extracellular potassium levels increase[90,91]. The latter lasts for roughly 10 min, during which the resting membrane potential decreases, approaching the firing threshold potential, thus accelerating electrical conduction[92]. The intracellular acidosis also drives an increase in cytosolic calcium, facilitating early and late depolarization, as well as spatial and temporal fluctuations in the duration of action potentials[93]. Additionally, ischemia leads to dephosphorylation of connexin-43 in gap junctions, which impairs intercellular electrical coupling and anisotropy[94]. Lastly, sympathetic stimulation not only promotes calcium release from the sarcoplasmic reticulum, but it also prompts lipolysis, elevating circulating free FA levels and therefore predisposing to ischemia-induced arrhythmogenesis[95].
Other possible mediators of cardiac arrhythmia include thrombin and endothelin-1. Patients with ST-elevation myocardial infarction (STEMI) complicated with ventricular fibrillation have been found to have higher levels of thrombin activity markers[96]. The production of thrombin at sites of coronary occlusion has been suggested to favor accumulation of lysophosphatidylcholine, which activates Na+ channels and Na+/H+ exchangers, enhancing the entry of Na+ into the cell[97]. On the other hand, endothelin-1 activity increases exponentially during acute ischemia, mediating direct and indirect electrophysiological effects through type-A endothelin receptors, and possibly playing a protecting role through type B endothelin receptors during the early stages of ischemia[98].
Acute pro-inflammatory cytokines - including TNF-α, IL-1β, IL-6 - and metalloproteinases are also found in greater proportions in the myocardium following an AMI, and may account for the electrophysiological changes observed in the cardiomyocytes within the border zone of the infarction[99]. This suggests an inflammatory process is involved in the remodeling and arrhythmogenic changes seen after an infarction. Patients with post-AMI arrhythmias tend to display higher levels of pro-inflammatory cytokines than those without arrhythmias[100,101]; and subjects without AMI or structural myocardial damage with systemic inflammation have shown an increased risk of ventricular tachyarrhythmias[102].
This catalogue of arrhythmogenic changes occur heterogeneously along the penumbra of the infarction zone, impairing its functionality as a syncytium and the electrical coupling among cardiomyocytes[103,104]. These phenomena are complex and appear to occur erratically due to the dynamic nature of reperfusion, low pH, oxygen depletion, increased intracellular calcium and potentiated sympathetic signaling[105]. These events, along with the high proportion of electrical decoupling and the auto-oscillatory activity of individual cells lay the foundation for a very variable syncytial behavior[106]. This leads to the continuous generation of mini-reentry circuits from individual ectopic rhythmic foci within the decoupled cell areas, which then spread to the better coupled zones within the penumbra, constituting the genesis of arrhythmias[107]. In concert, these changes are thought to be the triggers and substrates for ventricular arrhythmias, although further research is required to elucidate them in more detail.
Lipotoxic phenomena in ischemia and arrhythmia
In 1968, Oliver et al.[108] determined patients with AMI and especially those with elevated free FA levels had a greater incidence of ventricular arrhythmias in comparison with normolipemic patients. This finding has been also been found by various authors ever since[109]. More recently, in the Paris Prospective Study - a research realized in 5,250 male subjects followed during 22 years - high plasma free FA levels have been associated with sudden cardiac death, yet appeared to be unrelated to other causes of AMI[110].
The exact mechanism through which the shifts in FA/glucose metabolism in the cardiomyocyte lead to arrhythmia remain unclear, yet various alternatives have been proposed[111]. During an infarction, numerous events develop in parallel, with some of the earliest being precordial pain and increased sympathetic activity[112]. Although a moderate increase in catecholamines may aid in maintaining cardiac contractility in the face of oxygen depletion, excessive signaling can augment the energetic demands of the myocardium, impairing functionality[113].
It has been proposed that for the ischemic cardiomyocyte, glucose metabolism is more beneficial, and FA metabolism is deleterious[114]. However, FA availability is much greater: catecholamines induce lipolysis in adipose tissue, abruptly rising circulating FA levels[115], and inhibit insulin signaling, reducing glucose entry into the myocardium[116]. Likewise, FA can inhibit glucose oxidation and potentiate oxygen consumption in ischemic myocardial areas, leading to a preferential utilization of FA over glucose during ischemia[117].
The mechanisms underlying FA-related toxicity appear to be fundamentally linked with adrenergic stimulation[118]. However, FA also exhibit direct arrhythmogenic activity. Even without ischemia, a sufficiently high free FA/albumin molar rate can inhibit β-oxidation, leading to accumulation of acyl-carnitine and acyl-CoA in the cytosol. In turn, acyl-carnitine inhibits Ca++ pumps in the sarcoplasmic reticulum, as well as the Na+/Ca++ exchanger and the Na+/K+ ATPase pump, finalizing in an overload of Ca++ in the cytosol[119].
Furthermore, other metabolic processes activated upon elevation of free FA levels, such as membrane lipid peroxidation, inhibition of β-oxidation, uncoupling of proteins in the mitochondrial respiratory chain, accumulation of CoA derivates and extracellular K+ with shortening of action potentials, are all harmful for the ischemic cardiomyocyte[120]. On this basis, strong arguments propose the reduction of circulating free FA as a therapeutic measure in AMI, although further evidence is required to encourage this practice. Regarding instrumentation, the glucose-insulin-potassium (GIK) infusion is a well-known and viable alternative, as it promotes glucose entry into the myocardium and inhibits lipolysis, with the metabolic benefits this implies in cardiomyocytes[121]. Despite early clinical trials failing to demonstrate the utility of GIK at decreasing sudden death during the first 3 days following an AMI[122], more recent reports, such as the IMMEDIATE study have shown better outcomes at a 1-year follow-up: in patients with STEMI, 1-year mortality and hospitalizations for heart failure decreased significantly[123]. Theoretically, drugs for rapid inhibition of lipolysis would yield better results, yet research efforts in this line remain scarce and poor.
Quick myocardial FA availability has been closely related with epicardial adipose tissue (EAT) or the epicardial fat pad which is found between the myocardium and the visceral pericardium. This isin close proximity with the coronary arteries and so facilitates the activity of anti-inflammatory and pro-inflammatory adipokines and FA in the myocardium and arterial walls[124,125]. The Framingham Heart Study[126] has identified the EAT volume as a predictor of atrial fibrillation - the most frequent arrhythmia in clinical practice - independent of other methods for the measurement of adiposity such as the body mass index. Other studies have reported the EAT volume to be associated with the prevalence and severity of atrial fibrillation[127].
The EAT has also been found to release pro-inflammatory messengers such as activin A, which induces the expression of the transforming growth factor β1 (TGF-β1) and several metalloproteinases. These are key regulators in the homeostasis of the extracellular matrix - in particular by modifying collagen fibers - and mediate the profibrotic effect of EAT in the atrial myocardium[128]. Many other inflammatory mediators secreted by EAT also intervene in the pathogenesis of atrial fibrillation, including TNF-α, IL-8, and the monocyte chemotactic protein 1 (MCP-1)[129].
Finally, increased EAT has been associated with fatty infiltration in the myocardium, which contributes to tissue disorganization and promotes an arrhythmia-prone environment[130]. At the same time, these deposits favor the proliferation of myofibroblasts and increase the amount of dystrophic cardiomyocytes[130]. In this context, the EAT has been proposed as a novel therapeutic target in the management of cardiovascular disease[125].
Conclusion
The cardiomyocyte is the protagonist cell in the heart which allows it to function as a pump. This capacity requires strict electrophysiological and mechanical control and large amounts of ATP. These energetic demands are covered by a dynamic selection of substrates, shifting between carbohydrates and FA depending on various intracellular and systemic circumstances. Myocardial metabolism has been historically described as the "lost child of cardiology"[131], as clinicians have rather focused on thrombolysis and angioplasty, and very little practical value has been obtained from its study. Nonetheless, the evidence suggesting metabolic causes for arrhythmias grants a "resuscitation" of this cause[120]. Indeed, further research is required in order to explore and exploit the therapeutic implications of myocardial metabolism.
Declarations
Authors’ contributionsArticle conception and design: M.S. Martínez, A. García
Acquisition and analysis of bibliographic information: M.S. Martínez, A. García, E. Luzardo, L.C. Olivar
Drafting of the manuscript: M.S. Martínez, M. Chávez-Castillo, J. Salazar
Critical revision and final approval: V. Bermúdez, M. Velasco, J.J. Rojas Quintero
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2017.
REFERENCES
1. D'Souza K, Nzirorera C, Kienesberger P. Lipid metabolism and signaling in cardiac lipotoxicity. Biochim Biophys Acta 2016;1861:1513-24.
2. Gillespie HS, Lin CC, Prutkin JM. Arrhythmias in structural heart disease. Curr Cardiol Rep 2014;16:510.
3. Burton RA, Lee P, Casero R, Garny A, Siedlecka U, Schneider JE, Kohl P, Grau V. Three-dimensional histology: tools and application to quantitative assessment of cell-type distribution in rabbit heart. Europace 2014;16 Suppl 4:iv86-95.
4. Maass K, Shekhar A, Lu J, Kang G, See F, Kim EE, Delgado C, Shen S, Cohen L, Fishman GI. Isolation and characterization of embryonic stem cell-derived cardiac Purkinje cells. Stem Cells 2015;33:1102-12.
5. Vigmond EJ, Stuyvers BD. Modeling our understanding of the His-Purkinje system. Prog Biophys Mol Biol 2016;120:179-88.
6. Yin Z, Ren J, Guo W. Sarcomeric protein isoform transitions in cardiac muscle: a journey to heart failure. Biochim Biophys Acta 2015;1852:47-52.
7. Hu LY, Ackermann MA, Kontrogianni-Konstantopoulos A. The sarcomeric M-region: a molecular command center for diverse cellular processes. Biomed Res Int 2015;2015:714197.
8. Katz A. Contractile proteins of the heart. Phys Rev 2016;50:63-158.
9. Gazta-aga L, Marchlinski F, Betensky B. Mechanisms of cardiac arrhythmias. Rev Esp Cardiol (Engl Ed) 2012;65:174-85.
10. Kleber AG, Saffitz JE. Role of the intercalated disc in cardiac propagation and arrhythmogenesis. Front Physiol 2014;5:404.
11. Kurtenbach S, Kurtenbach S, Zoidl G. Gap junction modulation and its implications for heart function. Front Physiol 2014;5:82.
12. Veeraraghavan R, Poelzing S, Gourdie RG. Intercellular electrical communication in the heart: a new, active role for the intercalated disk. Cell Commun Adhes 2014;21:161-7.
13. Rider O, Cox P, Tyler D, Clarke K, Neubauer S. Myocardial substrate metabolism in obesity. Int J Obes (Lond) 2012;37:972-9.
14. Carley AN, Taegtmeyer H, Lewandowski ED. Mechanisms linking energy substrate metabolism to the function of the heart. Circ Res 2014;114:717-29.
15. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure - implications beyond ATP production. Circ Res 2013;113:709-24.
16. Rosano G, Vitale C, Spoletini I. Metabolic approach to heart failure: the role of metabolic modulators. Egyptian Heart J 2015;67:177-81.
17. Long Q, Yang K, Yang Q. Regulation of mitochondrial ATP synthase in cardiac pathophysiology. Am J Cardiovasc Dis 2015;5:19-32.
19. Wang J, Guo T. Metabolic remodeling in chronic heart failure. J Zhejiang Univ Sci B 2013;14:688-95.
20. Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metabolism 2012;15:805-12.
21. Wolf P, Winhofer Y, Krššák M, Krebs M. Heart, lipids and hormones. Endocr Connect 2017;6:R59-69.
22. Abumrad NA, Goldberg IJ. CD36 actions in the heart: lipids, calcium, inflammation, repair and more? Biochim Biophys Acta 2016;1860:1442-9.
23. Fukushima A, Lopaschuk GD. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim Biophys Acta 2016;1861:1525-34.
24. Fukushima A, Milner K, Gupta A, Lopaschuk GD. Acetylation control of cardiac fatty acid β-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim Biophys Acta 2016;1862:2211-20.
25. Fillmore N, Mori J, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol 2014;171:2080-90.
26. Glatz JF, Nabben M, Heather LC, Bonen A, Luiken JJ. Regulation of the subcellular trafficking of CD36, a major determinant of cardiac fatty acidutilization. Biochim Biophys Acta 2016;1861:1461-71.
27. Pepino M, Kuda O, Samovski D, Abumrad N. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Ann Rev Nutr 2014;34:281-303.
28. Glatz JF, Luiken JJ. From fat to FAT (CD36/SR-B2): understanding the regulation of cellular fatty acid uptake. Biochimie 2017;136:21-6.
29. Chanda D, Luiken JJ, Glatz JF. Signaling pathways involved in cardiac energy metabolism. FEBS Lett 2016;590:2364-74.
30. Kim TT, Dyck JR. The role of CD36 in the regulation of myocardial lipid metabolism. Biochim Biophys Acta 2016;1861:1450-60.
32. Glatz JF, Angin Y, Steinbusch LK, Schwenk RW, Luiken JJ. CD36 as a target to prevent cardiac lipotoxicity and insulin resistance. Prostaglandins Leukot Essent Fatty Acids 2013;88:71-7.
33. Samovski D, Su X, Xu Y, Abumrad N, Stahl P. Insulin and AMPK regulate FA translocase/CD36 plasma membrane recruitment in cardiomyocytes via Rab GAP AS160 and Rab8a RabGTPase. J Lipid Res 2012;53:709-17.
34. Angin Y, Steinbusch L, Simons P, Greulich S, Hoebers N, Douma K, van Zandvoort MA, Coumans WA, Wijnen W, Diamant M, Ouwens DM, Glatz JF, Luiken JJ. CD36 inhibition prevents lipid accumulation and contractile dysfunction in rat cardiomyocytes. Biochem J 2012;448:43-53.
35. Tate M, Grieve D, Ritchie R. Are targeted therapies for diabetic cardiomyopathy on the horizon? Clin Sci 2017;131:897-915.
36. Bairwa S, Parajuli N, Dyck J. The role of AMPK in cardiomyocyte health and survival. Biochim Biophys Acta 2016;1862:2199-210.
37. Vázquez-Carrera M. Unraveling the effects of PPARβ/δ on insulin resistance and cardiovascular disease. Trends Endocrinol Metab 2016;27:319-34.
38. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev 2010;90:207-58.
40. Barlaka E, Galatou E, Mellidis K, Ravingerova T, Lazou A. Role of pleiotropic properties of peroxisome proliferator-activated receptors in the heart: focus on the nonmetabolic effects in cardiac protection. Cardiovasc Ther 2016;34:37-48.
41. Barlaka E, Ledvényiová V, Galatou E, Ferko M, Čarnická S, Ravingerová T, Lazou A. Delayed cardioprotective effects of WY-14643 are associated with inhibition of MMP-2 and modulation of Bcl-2 family proteins through PPAR-α activation in rat hearts subjected to global ischaemia-reperfusion. Can J Physiol Pharmacol 2013;91:608-16.
42. Sun W, Liu Q, Leng J, Zheng Y, Li J. The role of pyruvate dehydrogenase complex in cardiovascular diseases. Life Sci 2015;121:97-103.
43. Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Asp Med 2013;34:121-38.
44. Deng D, Yan N. GLUT, SGLT, and SWEET: structural and mechanistic investigations of the glucose transporters. Protein Sci 2016;25:546-58.
45. Szablewski L. Glucose transporters in healthy heart and in cardiac disease. Int J Cardiol 2017;230:70-5.
46. Azevedo PS, Minicucci MF, Santos PP, Paiva SA, Zornoff LA. Energy metabolism in cardiac remodeling and heart failure. Cardiol Rev 2013;21:135-40.
47. Lopaschuk GD. Metabolic modulators in heart disease: past, present, and future. Can J Cardiol 2017;33:838-49.
48. Fillmore N, Lopaschuk GD. Targeting mitochondrial oxidative metabolism as an approach to treat heart failure. Biochim Biophys Acta 2013;1833:857-65.
49. Luptak I, Yan J, Cui L, Jain M, Liao R, Tian R. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation 2007;116:901-9.
50. Liao R, Jain M, Cui L, D'Agostino J, Aiello F, Luptak I, Ngoy S, Mortensen RM, Tian R. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 2002;106:2125-31.
51. Domenighetti AA, Danes VR, Curl CL, Favaloro JM, Proietto J, Delbridge LM. Targeted GLUT-4 deficiency in the heart induces cardiomyocyte hypertrophy and impaired contractility linked with Ca(2+) and proton flux dysregulation. J Mol Cell Cardiol 2010;48:663-72.
52. Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation 2007;116:434-48.
53. Varma N, Eberli FR, Apstein CS. Increased diastolic chamber stiffness during demand ischemia: response to quick length change differentiates rigor-activated from calcium-activated tension. Circulation 2000;101:2185-92.
54. Guimarães-Ferreira L. Role of the phosphocreatine system on energetic homeostasis in skeletal and cardiac muscles. Einstein 2014;12:126-31.
55. Wallimann T, Dolder M, Schlattner U, Eder M, Hornemann T, O'Gorman E, Rück A, Brdiczka D. Some new aspects of creatine kinase (CK): compartmentation, structure, function and regulation for cellular and mitochondrial bioenergetics and physiology. Biofactors 1998;8:229-34.
56. Weiss R, Gerstenblith G, Bottomley P. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Nat Acad Sci U S A 2005;102:808-13.
57. Fowler ED, Benoist D, Drinkhill MJ, Stones R, Helmes M, Wüst RC, Stienen GJ, Steele DS, White E. Decreased creatine kinase is linked to diastolic dysfunction in rats with right heart failure induced by pulmonary artery hypertension. J Mol Cell Cardiol 2015;86:1-8.
58. Bottomley PA, Panjrath GS, Lai S, Hirsch GA, Wu K, Najjar SS, Steinberg A, Gerstenblith G, Weiss RG. Metabolic rates of ATP transfer through creatine kinase (CK Flux) predict clinical heart failure events and death. Sci Transl Med 2013;5:215re3.
59. Kristjansson RP, Oddsson A, Helgason H, Sveinbjornsson G, Arnadottir GA, Jensson BO, Jonasdottir A, Jonasdottir A, Bragi Walters G, Sulem G, Oskarsdottir A, Benonisdottir S, Davidsson OB, Masson G, Magnusson OT, Holm H, Sigurdardottir O, Jonsdottir I, Eyjolfsson GI, Olafsson I, Gudbjartsson DF, Thorsteinsdottir U, Sulem P, Stefansson K. Common and rare variants associating with serum levels of creatine kinase and lactate dehydrogenase. Nat Commun 2016;7:10572.
60. Ventura-Clapier R, Garnier A, Veksler V. Energy metabolism in heart failure. J Physiol 2003;555:1-13.
61. Kitzenberg D, Colgan SP, Glover LE. Creatine kinase in ischemic and inflammatory disorders. Clin Transl Med 2016;5:31.
62. Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, Pabst T, Ertl G, Hahn D, Ingwall JS, Kochsiek K. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation 1997;96:2190-6.
63. Nakae I, Mitsunami K, Omura T, Yabe T, Tsutamoto T, Matsuo S, Takahashi M, Morikawa S, Inubushi T, Nakamura Y, Kinoshita M, Horie M. Proton magnetic resonance spectroscopy can detect creatine depletion associated with the progression of heart failure in cardiomyopathy. J Am Coll Cardiol 2003;42:1587-93.
64. Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 2009;297:E578-91.
65. Fukushima A, Milner K, Gupta A, Lopaschuk GD. Myocardial energy substrate metabolism in heart failure: from pathways to therapeutic targets. Curr Pharm Des 2015;21:3654-64.
66. Aroor A, Mandavia C, Sowers J. Insulin resistance and heart failure: molecular mechanisms. Heart Fail Clin 2012;8:609-17.
67. Breckenridge RA, Piotrowska I, Ng KE, Ragan TJ, West JA, Kotecha S, Towers N, Bennett M, Kienesberger PC, Smolenski RT, Siddall HK, Offer JL, Mocanu MM, Yelon DM, Dyck JR, Griffin JL, Abramov AY, Gould AP, Mohun TJ. Hypoxic regulation of hand1 controls the fetal-neonatal switch in cardiac metabolism. PLoS Biol 2013;11:e1001666.
68. Wende AR, Brahma MK, McGinnis GR, Young ME. Metabolic origins of heart failure. JACC Basic Transl Sci 2017;2:297-310.
69. Wende AR, Kim J, Holland WL, Wayment BE, O'Neill BT, Tuinei J, Brahma MK, Pepin ME, McCrory MA, Luptak I, Halade GV, Litwin SE, Abel ED. Glucose transporter 4 (GLUT4) deficient hearts develop maladaptive hypertrophy in response to physiologic or pathologic stresses. Am J Physiol Heart Circ Physiol 2017;313:H1098-108.
70. Wang J, Li Z, Wang Y, Zhang J, Zhao W, Fu M, Han X, Zhou J, Ge J. Qiliqiangxin enhances cardiac glucose metabolism and improves diastolic function in spontaneously hypertensive rats. Evid Based Complement Alternat Med 2017;2017:3197320.
71. Korvald C, Elvenes OP, Myrmel T. Myocardial substrate metabolism influences left ventricular energetics in vivo. Am J Physiol Heart Circ Physiol 2000;278:H1345-51.
72. Nagoshi T, Yoshimura M, Rosano GM, Lopaschuk GD, Mochizuki S. Optimization of cardiac metabolism in heart failure. Curr Pharm Des 2011;17:3846-53.
73. Babalis D, Tritakis V, Floros G, Mouzarou A, Kafkas N, Bampali K, Mertzanos G. Effects of ranolazine on left ventricular diastolic and systolic function in patients with chronic coronary disease and stable angina. Hellenic J Cardiol 2015;56:237-41.
74. Burkhoff D, Weiss RG, Schulman SP, Kalil-Filho R, Wannenburg T, Gerstenblith G. Influence of metabolic substrate on rat heart function and metabolism at different coronary flows. Am J Physiol 1991;261:H741-50.
75. Sabbah HN, Gupta RC, Rastogi S, Wang M. Dysregulation of mitochondria fission and fusion proteins in explanted failure human hearts. J Heart Lung Transplant 2011;30:S137.
76. Carolo dos Santos K, Pereira Braga C, Octavio Barbanera P, Seiva FR, Fernandes Junior A, Fernandes AA. Cardiac energy metabolism and oxidative stress biomarkers in diabetic rat treated with resveratrol. PLoS One 2014;9:e102775.
77. Maulucci G, Daniel B, Cohen O, Avrahami Y, Sasson S. Hormetic and regulatory effects of lipid peroxidation mediators in pancreatic beta cells. Mol Aspects Med 2016;49:49-77.
78. Ayala A, Mu-oz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014;2014:360438.
79. Gruzdeva O, Uchasova E, Dyleva Y, Belik E, Kashtalap V, Barbarash O. Relationship between free fatty acids, insulin resistance markers, and oxidized lipoproteins in myocardial infarction and acute left ventricular failure. Diabetes Metab Syndr Obes 2013;6:103-11.
80. Roy VK, Kumar A, Joshi P, Arora J, Ahanger AM. Plasma free fatty acid concentrations as a marker for acute myocardial infarction. J Clin Diagn Res 2013;7:2432-4.
81. Ma P, Han L, Lv Z, Chen W, Hu H, Tu J, Zhou X, Liu SM. In-hospital free fatty acids levels predict the severity of myocardial ischemia of acute coronary syndrome. BMC Cardiovasc Disord 2016;16:29.
82. Styskal J, Van Remmen H, Richardson A, Salmon AB. Oxidative stress and diabetes: what can we learn about insulin resistance from antioxidant mutant mouse models? Free Radic Biol Med 2012;52:46-58.
83. Abel ED, O'Shea KM, Ramasamy R. Insulin resistance: metabolic mechanisms and consequences in the heart. Arterioscler Thromb Vasc Biol 2012;32:2068-76.
84. Drosatos K, Schulze PC. Cardiaclipotoxicity: molecular pathways and therapeutic implications. Curr Heart Fail Rep 2013;10:109-21.
85. Park TS, Goldberg IJ. Sphingolipids, lipotoxic cardiomyopathy, and cardiac failure. Heart Fail Clin 2012;8:633-41.
86. Steggall A, Mordi IR, Lang CC. Targeting metabolic modulation and mitochondrial dysfunction in the treatment of heart failure. Diseases 2017;5:14.
87. Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation 2012;125:620-37.
88. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res 2015;116:1887-906.
89. Bhar-Amato J, Davies W, Agarwal S. Ventricular arrhythmia after acute myocardial infarction: "the perfect storm". Arrhythm Electrophysiol Rev 2017;6:134-9.
90. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 2012;298:229-317.
91. Gewirtz H, Dilsizian V. Myocardial viability: survival mechanisms and molecular imaging targets in acute and chronicischemia. Circ Res 2017;120:1197-212.
92. Baumeister P, Quinn TA. Altered calcium handling and ventricular arrhythmias in acute ischemia. Clin Med Insights Cardiol 2016;10:61-9.
93. Bai J, Yin R, Wang K, Zhang H. Mechanisms underlying the emergence of post-acidosis arrhythmia at the tissue level: a theoretical study. Front Physiol 2017;8:195.
94. Kolettis T. Coronary artery disease and ventricular tachyarrhythmia: pathophysiology and treatment. Curr Opin Pharmacol 2013;13:210-7.
95. Thackeray JT, Beanlands RS, DaSilva JN. Altered sympathetic nervous system signaling in the diabetic heart: emerging targets for molecular imaging. Am J Nucl Med Mol Imaging 2012;2:314-34.
96. Oikonomidis DL, Tsalikakis DG, Baltogiannis GG, Tzallas AT, Xourgia X, Agelaki MG, Megalou AJ, Fotopoulos A, Papalois A, Kyriakides ZS, Kolettis TM. Endothelin-B receptors and ventricular arrhythmogenesis in the rat model of acute myocardial infarction. Basic Res Cardiol 2009;105:235-45.
97. Francis Stuart S, De Jesus N, Lindsey M, Ripplinger C. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J Mol Cell Cardiol 2016;91:114-22.
98. Skovsted GF, Kruse LS, Berchtold LA, Grell AS, Warfvinge K, Edvinsson L. Myocardial ischemia-reperfusion enhances transcriptional expression of endothelin-1 and vasoconstrictor ETB receptors via the protein kinase MEK-ERK1/2 signaling pathway in rat. PLoS One 2017;12:e0174119.
99. Streitner F, Kuschyk J, Veltmann C, Ratay D, Schoene N, Streitner I, Brueckmann M, Schumacher B, Borggrefe M, Wolpert C. Role of proinflammatory markers and NT-proBNP in patients with an implantable cardioverter-defibrillator and an electrical storm. Cytokine 2009;47:166-72.
100. Maradit-Kremers H, Crowson CS, Nicola PJ, Ballman KV, Roger VL, Jacobsen SJ, Gabriel SE. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum 2005;52:402-11.
101. Downar E, Janse MJ, Durrer D. The effects of acute coronary artery occlusion on subepicardial transmembrane potentials in the intact porcine heart. Circulation 1977;56:217-24.
102. Biktashev VN, Biktasheva IV, Sarvazyan NA. Evolution of spiral and scroll waves of excitation in a mathematical model of ischaemic border zone. PLoS One 2011;6:e24388.
103. Solovyova O, Katsnelson L, Kohl P, Panfilov A, Tsaturyan A, Tsyvian P. Mechano-electric heterogeneity of the myocardium as a paradigm of its function. Prog Biophys Mol Biol 2016;120:249-54.
104. Robbers LF, Delewi R, Nijveldt R, Hirsch A, Beek AM, Kemme MJ, van Beurden Y, van der Laan AM, van der Vleuten PA, Tio RA, Zijlstra F, Piek JJ, van Rossum AC. Myocardial infarct heterogeneity assessment by late gadolinium enhancement cardiovascular magnetic resonance imaging shows predictive value for ventricular arrhythmia development after acute myocardial infarction. Eur Heart J Cardiovasc Imaging 2013;14:1150-8.
105. Pertsov AM, Davidenko JM, Salomonsz R, Baxter WT, Jalife J. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. Circ Res 1993;72:631-50.
106. Oliver MF. Control of free fatty acids during acute myocardial ischaemia. Heart 2010;96:1883-4.
108. Oliver MF, Kurien VA, Greenwood TW. Relation between serum free fatty acids and arrhythmias and death after acute myocardial infarction. Lancet 1968;1:710-4.
109. Tansey MJ, Opie LH. Relation between plasma free fatty acids and arrhythmias within the first twelve hours of acute myocardial infarction. Lancet 1983;2:419-22.
110. Ouven X, Charles MA, Desnos M, Ducimetiere P. Circulating non-esterified fatty acid level as a predictive risk factor for sudden death in the population. Circulation 2001;104:756-61.
111. Yang KC, Kyle JW, Makielski JC, Dudley SC. Mechanisms of sudden cardiac death: oxidants and metabolism. Circ Res 2015;116:1937-55.
112. Leach A, Fisher M. Myocardial ischaemia and cardiac pain - a mysterious relationship. Br J Pain 2013;7:23-30.
113. Ostrowski SR, Pedersen SH, Jensen JS, Mogelvang R, Johansson PI. Acute myocardial infarction is associated with endothelial glycocalyx and cell damage and a parallel increase in circulating catecholamines. Crit Care 2013;17:R32.
114. Kalra BS, Roy V. Efficacy of metabolic modulators in ischemic heart disease: an overview. J Clin Pharmacol 2012;52:292-305.
115. Nielsen TS, Jessen N, Jørgensen JO, Møller N, Lund S. Dissecting adipose tissue lipolysis: molecular regulation and implications for metabolic disease. J Mol Endocrinol 2014;52:R199-222.
116. Oliver M. Fatty acids and the risk of death during acute myocardial ischaemia. Clin Sci 2015;128:349-55.
117. Fragasso G. Deranged cardiac metabolism and the pathogenesis of heart failure. Card Fail Rev 2016;2:8-13.
118. Geerling JJ, Boon MR, Kooijman S, Parlevliet ET, Havekes LM, Romijn JA, Meurs IM, Rensen PC. Sympathetic nervous system control of triglyceride metabolism: novel concepts derived from recent studies. J Lipid Res 2014;55:180-9.
119. Opie L. Metabolic management of acute myocardial infarction comes to the fore and extends beyond control of hyperglycemia. Circulation 2008;117:2172-7.
120. Miller N, Mjøs O, Oliver M. Relationship of epicardial ST segment elevation to the plasma free fatty acid/albumin ratio during coronary occlusion in dogs. Clin Sci 1976;51:209-13.
121. Selker HP, Udelson JE, Massaro JM, Ruthazer R, D'Agostino RB, Griffith JL, Sheehan PR, Desvigne-Nickens P, Rosenberg Y, Tian X, Vickery EM, Atkins JM, Aufderheide TP, Sayah AJ, Pirrallo RG, Levy MK, Richards ME, Braude DA, Doyle DD, Frascone RJ, Kosiak DJ, Leaming JM, Van Gelder CM, Walter GP, Wayne MA, Woolard RH, Beshansky JR. One-year outcomes of out-of-hospital administration of intravenous glucose, insulin, and potassium (GIK) in patients with suspected acute coronary syndromes (from the IMMEDIATE [Immediate Myocardial Metabolic Enhancement During Initial Assessment and Treatment in Emergency Care] Trial). Am J Cardiol 2014;113:1599-605.
122. Mamas MA, Neyses L, Fath-Ordoubadi F. A meta-analysis of glucose-insulin-potassium therapy for treatment of acute myocardial infarction. Exp Clin Cardiol 2010;15:e20-4.
123. Evans R, Hauton D. The role of triacylglycerol in cardiac energy provision. Biochim Biophys Acta 2016;1861:1481-91.
124. Salazar J, Luzardo E, Mejías JC, Rojas J, Ferreira A, Rivas-Ríos JR, Bermúdez V. Epicardial fat: physiological, pathological, and therapeutic implications. Cardiol Res Pract 2016;2016:1-15.
125. Thanassoulis G, Massaro JM, O'Donnell CJ, Hoffmann U, Levy D, Ellinor PT, Wang TJ, Schnabel RB, Vasan RS, Fox CS, Benjamin EJ. Pericardial fat is associated with prevalent atrial fibrillation: the framingham heart study. Circ Arrhythm Electrophysiol 2010;3:345-50.
126. Al Chekakie MO, Welles CC, Metoyer R, Ibrahim A, Shapira AR, Cytron J, Santucci P, Wilber DJ, Akar JG. Pericardial fat is independently associated with human atrial fibrillation. J Am Coll Cardiol 2010;56:784-8.
127. Hatem SN, Sanders P. Epicardial adipose tissue and atrial fibrillation. Cardiovasc Res 2014;102:205-13.
128. Venteclef N, Guglielmi V, Balse E, Gaborit B, Cotillard A, Atassi F, Amour J, Leprince P, Dutour A, Clément K, Hatem SN. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur Heart J 2014;36:795-805.
129. Tsao HM, Hu WC, Wu MH, Tai CT, Lin YJ, Chang SL, Lo LW, Hu YF, Tuan TC, Wu TJ, Sheu MH, Chang CY, Chen SA. Quantitative analysis of quantity and distribution of epicardial adipose tissue surrounding the left atrium in patients with atrial fibrillation and effect of recurrence after ablation. Am J Cardiol 2011;107:1498-503.
130. Samanta R, Pouliopoulos J, Thiagalingam A, Kovoor P. Role of adipose tissue in the pathogenesis of cardiac arrhythmias. Heart Rhythm 2016;13:311-20.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Martínez MS, García A, Luzardo E, Chávez-Castillo M, Olivar LC, Salazar J, Velasco M, Rojas Quintero JJ, Bermúdez V. Energetic metabolism in cardiomyocytes: molecular basis of heart ischemia and arrhythmogenesis. Vessel Plus 2017;1:130-41. http://dx.doi.org/10.20517/2574-1209.2017.34
AMA Style
Martínez MS, García A, Luzardo E, Chávez-Castillo M, Olivar LC, Salazar J, Velasco M, Rojas Quintero JJ, Bermúdez V. Energetic metabolism in cardiomyocytes: molecular basis of heart ischemia and arrhythmogenesis. Vessel Plus. 2017; 1: 130-41. http://dx.doi.org/10.20517/2574-1209.2017.34
Chicago/Turabian Style
Martínez, María Sofía, Andrés García, Eliana Luzardo, Mervin Chávez-Castillo, Luis Carlos Olivar, Juan Salazar, Manuel Velasco, Joselyn Joanna Rojas Quintero, Valmore Bermúdez. 2017. "Energetic metabolism in cardiomyocytes: molecular basis of heart ischemia and arrhythmogenesis" Vessel Plus. 1: 130-41. http://dx.doi.org/10.20517/2574-1209.2017.34
ACS Style
Martínez, MS.; García A.; Luzardo E.; Chávez-Castillo M.; Olivar LC.; Salazar J.; Velasco M.; Rojas Quintero JJ.; Bermúdez V. Energetic metabolism in cardiomyocytes: molecular basis of heart ischemia and arrhythmogenesis. Vessel Plus. 2017, 1, 130-41. http://dx.doi.org/10.20517/2574-1209.2017.34
About This Article
Copyright

Data & Comments
Data


 Cite This Article 122 clicks
Cite This Article 122 clicks

 Like This Article 30
likes
Like This Article 30
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.