
Perivascular mast cells promote neointimal elastin deposition and suppress chronic vein graft restenosis in hyperlipidaemic mice
 0
0
Abstract
Aim: Mast cells are versatile innate immune cells and are reported to promote vascular inflammation and neointimal lesion formation, thereby contributing to the development of vascular stenosis and atherosclerosis. However, it is not clear whether mast cells also regulate vascular matrix remodelling in established neointima. This study addressed the hypothesis that perivascular mast cells regulate neointimal matrix remodelling using a mouse vein graft model.
Methods: The impact of mast cells on neointimal remodelling was investigated using mast cell-deficient animals in both normolipidaemic (KitW-sh/W-sh) and hyperlipidaemic (apoE-/-KitW-sh/W-sh) conditions. The effect of perivascular mast cells on vascular matrix remodelling, including collagen and elastin deposition, was investigated using a local mast cell reconstitution method that selectively repopulated mast cells around the carotid artery (where the vein graft was inserted) in KitW-sh/W-sh mice.
Results: In normolipidaemic vein grafts (KitW-sh/W-sh vs. the wild type control C57BL/6J), collagen synthesis was not affected by mast cell deficiency at 4 weeks. In contrast, neointimal elastin was reduced by 6.5-fold in mast cell-deficient KitW-sh/W-sh mice, which was prevented by perivascular mast cell reconstitution. Mast cell deficiency induced a similar reduction in neointimal elastin in hyperlipidaemic mice (apoE-/-KitW-sh/W-sh vs. apoE-/-), with a significant increase in cell proliferation and neointimal area at 4 weeks.
Conclusion: Mast cells appear to promote elastin deposition in vein grafts and this may lead to further suppression of cell proliferation and neointimal thickening under hyperlipidaemic conditions.
Keywords
Introduction
Mast cells are versatile innate immune cells, well-known for their role in inflammation and innate immunity.[1,2] Despite the wide distribution of mast cells in arterial adventitia and perivascular connective tissue, our understanding of the influence of mast cells in vascular disease is limited. In recent years, accumulating evidence suggests that mast cells promote vascular inflammation and contribute to the progression of a number of vascular diseases including atherosclerosis, aortic aneurysm and vein graft neointima hyperplasia.[3-6] Systemic activation of mast cells using dinitrophenyl-albumin increased plaque size in apoE-/- mice, whilst selective stimulation of perivascular mast cells had no impact on plaque formation but destabilised established plaque with increased intra-plaque haemorrhage.[7] Compound 48/80, another mast cell activator, demonstrated a similar effect to promote atherosclerosis development, which was inhibited by the mast cell stabiliser cromolyn.[8,9] In addition to pharmacological manipulation of mast cell function, studies using genetic mast cell deficiency (as a consequence of spontaneous mutation in the promoter region of the c-kit gene[10]) also confirmed the detrimental effect of mast cells in atherosclerosis.[3] Mechanistic studies revealed that activated mast cells synthesised and released a wide range of pro-inflammatory factors including interleukin (IL)-6 and IL-8, interferon gamma, tumournecrosis factor alpha, histamine, chymase and tryptase. Consequently, mast cells exacerbated vascular inflammation with increased intra-plaque leukocyte infiltration, lipid uptake, vascular matrix degradation and subsequent plaque expansion and destabilisation.[6]
In vein graft disease, neointimal hyperplasia is the major cause of restenosis.[11,12] The neointima formation in vein grafts is mainly driven by acute vascular inflammation. In a mouse vein graft model, the acute inflammation usually lasts less than two weeks following grafting surgery.[13,14] The neointima thereafter undergoes a remodelling stage where neointimal cells differentiate into smooth muscle-like cells and synthesise large amount of vascular matrix including collagen and elastin as a process of arterialisation.[12,15] The remodelling process stabilises neointimal cells and prevents chronic neointimal thickening. We and others have found that perivascular mast cells boost acute vein graft inflammation via an increase in cytokine secretion and activation of the complement system.[14,16] This leads to a significant rise in cell proliferation in the acute inflammation stage (one week after vein graft implantation) and more neointima formation. However, it is not clear whether perivascular mast cells also regulate vascular matrix remodelling after resolution of acute inflammation. This is particularly important under pro-atherosclerotic conditions as vascular smooth muscle cells from pro-atherosclerotic animals are more susceptible to chronic proliferation in response to mitogenic stimuli.[17] In the present study, we performed interpositional vein grafting in mast cell deficient mouse lines bred on both a normolipidaemic and hyperlipidaemic genetic background (KitW-sh/W-sh and apoE-/-KitW-sh/W-sh) to address this question.
Methods
Animals
All experiments which involved animals conformed to Directive 2010/63/EU of the European Parliament and also to the UK Home Office Animal (Scientific Procedures) Act 1986 and were performed under Project Licence PPL 60/4114. In addition, ethical permission for the study had been granted by the University of Strathclyde ethical review committee.
Mice carrying the KitW-sh/W-sh mutation were used in this study as a mast cell deficiency model. KitW-sh/W-sh is a spontaneous mutation in the promoter region of the c-kit gene which is critical for mast cell survival.[10] As the c-kit gene itself is intact, c-kit expression is preserved to some extent in the early life of these mice. Consequently, systemic mast cell deficiency does not develop until 4 weeks old. Jackson Laboratories (USA) was the original supplier of both the KitW-sh/W-sh and apoE-/- mice and these were subsequently bred in-house. Both strains were on a C57BL/6J background. In order to generate a hyperlipidaemic mouse line lacking mast cells, apoE-/- and KitW-sh/W-sh mice were cross-bred. For the normolipidaemic KitW-sh/W-sh mice, the congenic control was C57BL/6J while for the hyperlipidaemic apoE-/-KitW-sh/W-sh mice, the apoE-/- was the congenic control. All mice were maintained on a cycle of 12-h periods of light and dark and allowed access to normal chow diet and water ad libitum. Male mice (about 10 to 20 weeks old) were used in this study.
Surgery
The mouse vein graft model is a well-established model for studying neointimal hyperplasia.[13,14] Briefly, i.p. injection of sodium pentobarbital (60 mg/kg) was used as an anaesthetic agent with top-up doses administered as appropriate and depending on the depth of anaesthesia demonstrated by the pedal withdrawal reflex. All animals received perioperative analgesic cover (buprenorphine; 0.05 mg/kg body weight, s.c.). From a donor mouse, the thoracic inferior vena cava was carefully harvested. In all experiments the donor mouse was a male mouse of the same genotype. In the recipient mouse, the right common carotid artery was prepared by isolating it, tying two sutures around the middle and cutting. A nylon cuff was then sleeved onto the distal arterial end. In order to graft the vein onto artery rather than nylon cuff, the artery was everted back over the cuff and ligated using 8/0 silk suture (Ethicon, Livingston, UK). The proximal end of the carotid artery was prepared in an identical fashion. The vena cava to be grafted was then sleeved onto each arterial end in turn and tied into position with 8/0 suture. Twenty-eight days after grafting, mice were euthanised by a rising concentration of CO2. The neck was opened and the vein graft removed and placed in physiological bathing solution.
Perivascular mast cell reconstitution
In a previous study from our laboratory, we established a reliable method for reconstituting mast cells locally to the perivascular region. The advantage of this method is that it re-establishes a local mast cell reconstitution in the KitW-sh/W-sh mice without mast cells being present elsewhere.[14] Thus, this method allows the role of perivascular mast cells to be investigated without the consequences of systemic mast cell reconstitution, which can include ectopic mast cell accumulation and abnormal distribution of mast cells in target organs/tissues.[10,14] Briefly, C57BL/6J mice were used as the source of bone marrow cells and these were cultured with mast cell-differentiating media (containing murine IL-3 and stem cell factor; PeproTech, New Jersey, USA) for 4 weeks as we have described previously.[14] To confirm that the cultured cells were in fact differentiated into mast cells, flow cytometry with anti-c-Kit and anti-FcεRI antibodies (eBioscience, Hatfield, UK) was used.[14] To investigate the impact of local reconstitution of mast cells on neointima formation within the vein graft, the perivascular area of the right common carotid artery was injected with the bone marrow-derived mast cells (BMMCs) as follows. Briefly, the recipient KitW-sh/W-sh mouse was anaesthetised with sodium pentobarbital as previously described and the common right carotid artery was exposed. BMMCs were injected around the artery (1 million cells per mouse). A suture of size 6/0 was used to close the wound and the animal was kept for 4 weeks to allow repopulation of the perivascular area with a mast cell population similar to that seen in wild type (C57BL/6) mice.[14] Vein graft surgery was performed 4 weeks after mast cell reconstitution.
Histology and immunostaining
Vein grafts were perfusion-fixed to maintain graft patency and then stored overnight in 10% formalin before being embedded in paraffin. For each vein graft, serial sections of 5 µm thickness were cut from 5 evenly divided regions from the proximal to the distal end. In all cases, 5 slides (1 slide from each region) were stained as outlined below and the values averaged for each vein graft. The advantage of this method is that the average value gives an indication of the value from the whole of the graft rather than one discrete area which may not be representative of the graft as a whole. For general and gross morphology and for planimetry studies, haematoxylin and eosin staining was used and the slides were photographed and analysed using Image pro plus software (Media Cybernetics, Marlow, UK). Collagen was stained by picrosirius red and visualised under polarised light. As with other stains used, at least 5 slides were used from the length of the graft to give a mean intensity of the total fluorescent signal and this value was used for comparison of collagen content between groups. Elastin was identified by Verhoeff Van Gieson staining and mean intensity of light absorption of the bluish dark stain within the neointima was quantified. To avoid batch-to-batch variation, for each staining protocol, all the vein graft samples were stained at one time. For immunostaining, slides were de-waxed and pressure-cooked in citrate buffer to retrieve antigens of interest. To study proliferation in vein grafts, one slide per vein graft was stained using an antibody to the marker Ki67 (rabbit anti-Ki67 antibody, Abcam). The percentage cell proliferation was presented as the ratio of Ki67 positive nuclei over total nuclei in the wall of the vein graft.
Statistics
In all experiments, data are presented as mean ± standard error of the mean where n refers to the number of mice. To make comparisons between two groups an unpaired Student's t-test was used as long as the data had a normal distribution and, for data not normally distributed, the non-parametric Mann Whitney test was used. To compare data from multiple groups a one-way analysis of variance (ANOVA) with Tukey's post hoc test was used as appropriate. Statistical analyses were performed using Graph Pad Prism v6.04. In all comparisons, a significant difference was assumed when P < 0.05.
Results
Mast cells promote elastin but not collagen deposition in vein grafts by 4 weeks
Significant collagen deposition was identified in all vein grafts at 4 weeks. The majority of collagen was present in the neo-adventitia and perivascular area [Figure 1A]. In contrast, elastin was only detected in the neointima [Figure 1B and C]. Mast cell deficiency (KitW-sh/W-sh) had no impact on collagen deposition, but suppressed elastin deposition by 6.5 fold. Local mast cell reconstitution (4 weeks before vein graft implantation) rescued the impaired elastin deposition in KitW-sh/W-sh vein grafts (23.5 ± 4.8 vs. 3.6 ± 1.1 vs. 26.4 ± 4.4 arbitrary units/mm2; C57BL/6J vs. KitW-sh/W-sh vs. KitW-sh/W-sh + lcMC; P < 0.01 by one-way ANOVA). Mast cell deficiency in hyperlipidaemic mice induced a similar suppression of neointimal elastin (37.6 ± 4.0 vs. 13.4 ± 2.9 arbitrary units/mm2; apoE-/- vs. apoE-/-KitW-sh/W-sh; P < 0.01 by Student’s t-test).
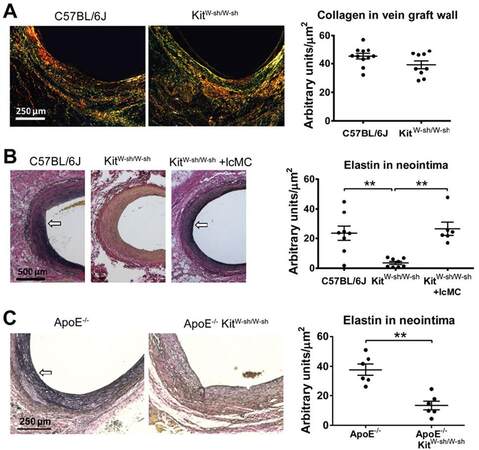
Figure 1. Mast cell deficiency suppressed elastin deposition in vein graft. (A) Picrosirus red staining demonstrated that both C57BL/6J and KitW-sh/W-sh vein grafts synthesised large amount of collagen (in green, yellow and red colour) in the adventitia and perivascular connective tissue by 4 weeks. No statistical difference was detected in collagen content between groups; (B and C) Elastin deposition was restricted to the vein graft neointima (visualised by Verhoeff Van Gieson staining, indicated by arrows). Mast cell deficient KitW-sh/W-sh vein grafts contained a significantly lower level of elastin versus congenic C57BL/6J vein grafts, which was rescued by local mast cell reconstitution. Mast cell deficiency in hyperlipidaemic mice (apoE-/-) also significantly suppressed elastin deposition in the vein graft by 4 weeks. n = 6; **P < 0.01. One-way analysis of variance plus Tukey post hoc test was used for multiple group comparison and unpaired student's t-test for two group comparison
Mast cell deficiency increased chronic neointimal proliferation and thickening in apoE-/- mice but not in C57BL/6J mice
Mast cell deficiency moderately suppressed neointima formation in normolipidaemic mice 4 weeks after grafting (1.7 × 105 ± 0.2 × 105 vs. 1.1 × 105 ± 0.1 × 105 µm2; C57BL/6J vs. KitW-sh/W-sh; P < 0.01 by Student’s t-test), but induced a 2-fold increase in the hyperlipidaemic mice (1.9 × 105 ± 0.2 × 105 vs. 4.2 × 105 ± 0.8 × 105 µm2; apoE-/- vs. apoE-/-KitW-sh/W-sh; P < 0.01 by Student’s t-test; Figure 2A and B). Cell proliferation within the vein graft at 4 weeks was very low in both C57BL/6J and KitW-sh/W-sh (2.1 ± 0.5% vs. 1.8 ± 0.3%; C57BL/6J vs. KitW-sh/W-sh), but significantly elevated in apoE-/-KitW-sh/W-sh compared to apoE-/- (1.9 ± 0.5% vs. 5.3 ± 1.4%; apoE-/- vs. apoE-/-KitW-sh/W-sh; P < 0.05 by Mann Whitney test; Figure 2A and C).
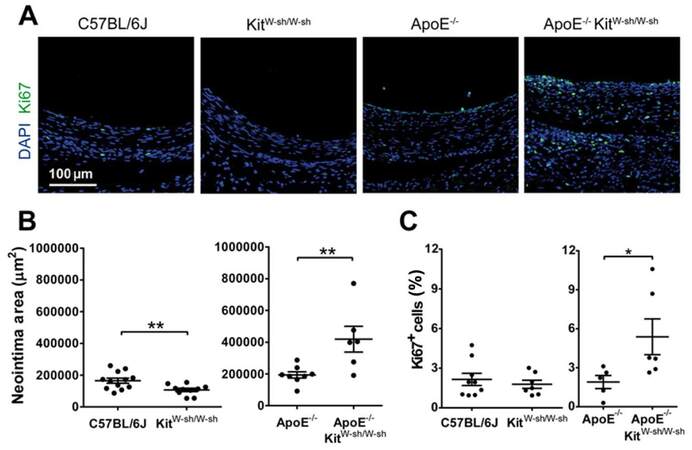
Figure 2. Mast cell deficiency in hyperlipidaemic mice increased neointima area with elevated chronic cell proliferation. (A) Representative images of cell proliferation in 4-week-old vein grafts. Proliferating cells were labelled using an antibody against Ki67 (green) and cell nuclei by DAPI (blue). The neointimal area in apoE-/- KitW-sh/W-sh vein grafts was significantly higher than all other groups (B), associated with elevated cell proliferation by 4 weeks (C). n > 6 for all groups; **P < 0.01 by one-way analysis of variance plus Tukey post hoc test
Discussion
This study demonstrates firstly that perivascular mast cells elevate neointimal elastin deposition under both normolipidaemic and hyperlipidaemic conditions, and secondly that they suppress neointimal thickening in hyperlipidaemic mice possibly via down regulation of cell proliferation within the vein graft.
Elastin is one of the fundamental structural proteins of the arterial wall that regulates vascular elasticity and stabilises smooth muscle cells.[18] The present study demonstrates that mast cells play a previously unrecognised role in promotion of elastin deposition during vein graft remodelling. The causality is demonstrated by the data showing that mast cell deficiency reduced, and mast cell reconstitution rescued, elastin deposition in the vein graft. Although the exact mechanism of how mast cells regulate elastin deposition is not yet clear, there is a consensus within the literature that heparin is the potential mediator. The evidence for the involvement of heparin is three-fold. Firstly, mast cells are the only cell type that produces heparin in vivo.[19] Secondly, heparin is known to promote elastogenesis and supress neointima hyperplasia in injured arteries and vascular grafts.[20,21] Being the most negatively charged molecule in biological systems, heparin covalently binds to the positively charged tropoelastin to accelerate tropoelastin coacervation and elastic fibre formation.[22-24] However, the very short half-life of heparin limits its pharmacological potency for therapeutic purposes.[25] Only continuous intravenous delivery,[26-28] but not short term treatment,[29] inhibited neointima thickening. Interestingly, perivascular delivery of heparin required a much smaller dose and was more effective in suppression of neointima hyperplasia,[20,25] which matches the source of endogenous heparin from perivascular mast cells. Thirdly, mast cell granules are enriched with heparin and the heparin-based particles are capable of long distance travel within tissue[30] which makes perivascular mast cells an ideal and indeed the only source for continuous heparin supply to assist vascular elastogenesis.
It is intriguing that mast cell-dependent elastin deposition had a divergent impact on neointimal hyperplasia in normolipidaemic and hyperlipidaemic vein grafts. This could be a consequence of the different dynamics of neointima formation and vascular matrix remodelling. In normolipidaemic mice, the neointima formation is driven by acute inflammation and proliferation which peak within one week and are complete by 2 weeks.[13,31] During the 3rd and 4th weeks, the neointimal proliferation decreases over 10-fold and the majority of the neointimal cells transdifferentiate into smooth muscle-like cells that initiate vascular matrix remodelling including elastin deposition.[13,14,18,31] Therefore, elastin is unlikely to regulate the early events of neointima formation in the normolipidaemic vein graft. The modest reduction in neointimal area in the KitW-sh/W-sh vein graft is attributed to the damped cell proliferation during the first week rather than altered elastin deposition.[14]
Both our data and the literature[32] demonstrated that apoE-/- mice, when fed on normal chow diet, do not demonstrate any enhancement of neointimal hyperplasia. The low cell proliferation level in apoE-/- vein grafts at 4 weeks in the current study suggests that the neointima was stabilised, similar to that in C57BL/6J. These observations are in contrast with in vitro evidence that apoE-/- smooth muscle cells are predisposed to a higher rate of proliferation.[17] One clear difference between data from cell cultures and neointima formation in vivo is the absence of the rich extracellular matrix environment. ApoE-/- vein grafts at 4 weeks contained large amounts of elastin that can stabilise neointimal smooth muscle cells.[18] The reduction in neointimal elastin in apoE-/-KitW-sh/W-sh grafts was correlated with a dramatic increase in cell proliferation within the vein graft wall, suggesting that the presence of elastin is an important protective mechanism against chronic smooth muscle cell proliferation under pro-atherogenic conditions.
Mast cells, as innate immune cells, are well-documented to have pro-inflammatory effects in many pre-clinical vascular studies.[6]Induction of mast cell activation in a pro-inflammatory environment, either using pharmacological activators or long term western diet in pro-atherosclerotic mouse models (apoE-/- or LDLr-/-), was a common feature of these studies. The widely accepted paradigm is that high levels of pro-inflammatory factors and proteinases released by activated mast cells promote vascular inflammation and development of vascular lesions. Indeed activated mast cells may be the explanation for the clinical correlation between mast cell-mediated allergic disease and atherosclerosis.[33] However, the role of unstimulated mast cells in normal vasculature or in stable vascular disease is not fully understood. The current study provides the first evidence that mast cells could exert a positive effect in vascular remodelling via regulation of elastin deposition. Our unpublished data suggests that the expression of inflammatory cytokines in vein grafts during the remodelling stage (4 weeks after implantation) is negligible. We therefore speculate that the mast cell-dependent regulation of elastin is independent of immunological/inflammatory mechanisms, but mediated by heparin (as discussed above) or some unknown mechanisms.
The mast cell-elastin-axis could potentially be extrapolated to vascular development as the first appearance of mast cells around the foetal aorta is before the formation of aortic elastic laminae.[34] This suggests that mast cells may also contribute to the foetal arteriogenesis by assisting formation of elastic fibres. In addition to the perivascular area, the mast cell population is clearly enriched in all the elastic tissue/organs including arteries, skin and lung, indicating a universal link between mast cells and elastin. Under pathological conditions, an increase of mast cell number is often associated with elastin-related tissue fibrosis.[35,36] In a skin photodamage model, chronic ultraviolet light exposure increases skin mast cell number accompanied by a 3.6 fold elevation in elastin content, which was not observed in the mast cell-deficient KitW/W-v mice.[37] Similar correlation was also present in human skin.[38] Non-elastic organs, such as the liver usually hosts only a very small population of mast cells.[39] In liver disease, an increase of hepatic elastin is one of the most distinct features of hepatic fibrosis and cirrhosis,[36] correlated with a significant increase in hepatic mast cells.[39] In contrast to other leukocyte recruitment, the increase of mast cell number persisted with the chronic progress of hepatic cirrhosis,[40] suggesting the mast cell-mediated pathological elastin accumulation was beyond inflammatory mechanisms.
Collectively, accumulating evidence suggests that the mast cell-elastin axis may be a universal mechanism that regulates physiological and pathological elastin metabolism. Our findings, in line with other literature, raise the question of whether mast cells regulate vascular elastin metabolism via mechanisms independent from their immunological function. More comprehensive investigation is required to clarify the complex role of mast cells in vascular development and remodelling.
The current study used a mast cell-deficient mouse model (KitW-sh/W-sh) and local mast cell reconstitution to demonstrate that perivascular mast cells promoted neointimal elastin remodelling in vein grafts. Limited by the experimental period, it was not clear whether perivascular mast cells merely accelerated the elastin deposition in the 4-week time window, or permanently increased the elastin level in the neointima. Unlike the elastic lamina in healthy arteries, the elastic fibres in the neointima were diffuse. The presence of mast cells only increased the amount of elastic fibres but did not improve the structure of the matrix. Further experiments are required to clarify whether the quantitative increase in elastin content has a positive impact on vascular compliance and vasomotor function, which may also influence the neointima hyperplasia.
In conclusion, this is the first study to demonstrate the mast cell-dependent regulation of elastin deposition in vein grafts, and subsequent suppression of chronic vein graft hyperplasia in hyperlipidaemic mice. This study sheds new light on the function of perivascular mast cells on vascular lesion formation and remodelling, and provides new targets for therapeutic intervention for vein graft failure and other stenotic vascular disease.
Declarations
Authors’ contributionsPerforming all the experiments, acquiring and analysing the data and writing the first draft of the paper: J. Wu
Designing the experiments, manuscript editing and reviewing: C. Lawrence, R.M. Wadsworth, S. Kennedy
Financial support and sponsorshipThis work was supported by a British Heart Foundation project grant (PG/09/095). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalThe study had been granted by the University of Strathclyde ethical review committee.
REFERENCES
1. St John AL, Abraham SN. Innate immunity and its regulation by mast cells. J Immunol 2013;190:4458-63.
2. Kennedy S, Wu J, Wadsworth RM, Lawrence CE, Maffia P. Mast cells and vascular diseases. Pharmacol Ther 2013;138:53-65.
3. Sun J, Sukhova GK, Wolters PJ, Yang M, Kitamoto S, Libby P, MacFarlane LA, Mallen-St Clair J, Shi GP. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med 2007;13:719-24.
4. Zhang J, Sun J, Lindholt JS, Sukhova GK, Sinnamon M, Stevens RL, Adachi R, Libby P, Thompson RW, Shi GP. Mast cell tryptase deficiency attenuates mouse abdominal aortic aneurysm formation. Circ Res 2011;108:1316-27.
5. de Vries MR, Wezel A, Schepers A, van Santbrink PJ, Woodruff TM, Niessen HW, Hamming JF, Kuiper J, Bot I, Quax PH. Complement factor C5a as mast cell activator mediates vascular remodelling in vein graft disease. Cardiovasc Res 2013;97:311-20.
6. Bot I, Shi GP, Kovanen PT. Mast cells as effectors in atherosclerosis. Arterioscler Thromb Vasc Biol 2015;35:265-71.
7. Bot I, de Jager SC, Zernecke A, Lindstedt KA, van Berkel TJ, Weber C, Biessen EA. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation 2007;115:2516-25.
8. Wang J, Sjöberg S, Tia V, Secco B, Chen H, Yang M, Sukhova GK, Shi GP. Pharmaceutical stabilization of mast cells attenuates experimental atherogenesis in low-density lipoprotein receptor-deficient mice. Atherosclerosis 2013;229:304-9.
9. Tang Y, Yang Y, Wang S, Huang T, Tang C, Xu Z, Sun YH. Mast cell degranulator compound 48-80 promotes atherosclerotic plaque in apolipoprotein E knockout mice with perivascular common carotid collar placement. Chin Med J (Engl) 2009;122:319-25.
10. Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol 2005;167:835-48.
11. Wu J, Zhang C. Neointimal hyperplasia, vein graft remodeling, and long-term patency. Am J Physiol Heart Circ Physiol 2009;297:H1194-5.
13. Wu J, Wadsworth RM, Kennedy S. Inhibition of inducible nitric oxide synthase promotes vein graft neoadventitial inflammation and remodelling. J Vasc Res 2011;48:141-9.
14. Wu J, Grassia G, Cambrook H, Ialenti A, MacRitchie N, Carberry J, Wadsworth RM, Lawrence C, Kennedy S, Maffia P. Perivascular mast cells regulate vein graft neointimal formation and remodeling. Peer J 2015;3:e1192.
15. de Vries MR, Simons KH, Jukema JW, Braun J, Quax PH. Vein graft failure: from pathophysiology to clinical outcomes. Nat Rev Cardiol 2016;13:451-70.
16. de Vries MR, Wezel A, Schepers A, van Santbrink PJ, Woodruff TM, Niessen HW, Hamming JF, Kuiper J, Bot I, Quax PH. Complement factor C5a as mast cell activator mediates vascular remodelling in vein graft disease. Cardiovasc Res 2013;97:311-20.
17. Kothapalli D, Castagnino P, Rader DJ, Phillips MC, Lund-Katz S, Assoian RK. Apolipoprotein E-mediated cell cycle arrest linked to p27 and the Cox2-dependent repression of miR221/222. Atherosclerosis 2013;227:65-71.
18. Lannoy M, Slove S, Jacob MP. The function of elastic fibers in the arteries: beyond elasticity. Pathol Biol 2014;62:79-83.
19. Carlsson P, Kjellén L. Heparin biosynthesis. In: Lever R, Mulloy B, Page CP, editors. Handbook of experimental pharmacology. Berlin, Heidelberg: Springer Berlin Heidelberg; 2012. pp. 23-41.
20. Teomim D, Fishbien I, Golomb G, Orloff L, Mayberg M, Domb AJ. Perivascular delivery of heparin for the reduction of smooth muscle cell proliferation after endothelial injury. J Control Release 1999;60:129-42.
21. Saitow C, Kaplan DL, Castellot JJ. Heparin stimulates elastogenesis: application to silk-based vascular grafts. Matrix Biol 2011;30:346-55.
22. Tu Y, Weiss AS. Transient tropoelastin nanoparticles are early-stage intermediates in the coacervation of human tropoelastin whose aggregation is facilitated by heparan sulfate and heparin decasaccharides. Matrix Biol 2010;29:152-9.
23. Cain SA, Baldock C, Gallagher J, Morgan A, Bax DV, Weiss AS, Shuttleworth CA, Kielty CM. Fibrillin-1 interactions with heparin: implications for microfibril and elastic fiber assembly. J Biol Chem 2005;280:30526-37.
24. Tu Y, Weiss AS. Glycosaminoglycan-mediated coacervation of tropoelastin abolishes the critical concentration, accelerates coacervate formation, and facilitates spherule fusion: implications for tropoelastin microassembly. Biomacromolecules 2008;9:1739-44.
25. Edelman ER, Adams DH, Karnovsky MJ. Effect of controlled adventitial heparin delivery on smooth muscle cell proliferation following endothelial injury. Proc Natl Acad Sci U S A 1990;87:3773-7.
26. Capron L, Jarnet J, Heudes D, Joseph-Monrose D, Bruneval P. Repeated balloon injury of rat aorta: a model of neointima with attenuated inhibition by heparin. Arterioscler Thromb Vasc Biol 1997;17:1649-56.
27. Chajara A, Heudes D, Peronneau I, Jarnet J, Basset A, Capron L. Effects of heparinoids on the sclerotic reaction of rat thoracic aorta to injury: comparison between standard and low-molecular-weight heparins in vitro and in vivo. J Cardiovasc Pharmacol 1994;23:995-1003.
28. Guyton JR, Rosenberg RD, Clowes AW, Karnovsky MJ. Inhibition of rat arterial smooth muscle cell proliferation by heparin. In vivo studies with anticoagulant and nonanticoagulant heparin. Circ Res 1980;46:625-34.
29. Ellis SG, Roubin GS, Wilentz J, Douglas JS, King SB. Effect of 18- to 24-hour heparin administration for prevention of restenosis after uncomplicated coronary angioplasty. Am Heart J 1989;117:777-82.
30. Kunder CA, St John AL, Li G, Leong KW, Berwin B, Staats HF, Abraham SN. Mast cell-derived particles deliver peripheral signals to remote lymph nodes. J Exp Med 2009;206:2455-67.
31. Marx SO, Totary-Jain H, Marks AR. Vascular smooth muscle cell proliferation in restenosis. Circ Cardiovasc Interv 2011;4:104-11.
32. Matter CM, Ma L, von Lukowicz T, Meier P, Lohmann C, Zhang D, Kilic U, Hofmann E, Ha SW, Hersberger M, Hermann DM, Lüscher TF. Increased balloon-induced inflammation, proliferation, and neointima formation in apolipoprotein E (ApoE) knockout mice. Stroke 2006;37:2625-32.
33. Liu CL, Zhang JY, Shi GP. Interaction between allergic asthma and atherosclerosis. Transl Res 2016;174:5-22.
34. Wulff BC, Parent AE, Meleski MA, DiPietro LA, Schrementi ME, Wilgus TA. Mast cells contribute to scar formation during fetal wound healing. J Invest Dermatol 2012;132:458-65.
35. Wilgus TA, Wulff BC. The importance of mast cells in dermal scarring. Adv Wound Care (New Rochelle) 2014;3:356-65.
36. Pellicoro A, Aucott RL, Ramachandran P, Robson AJ, Fallowfield JA, Snowdon VK, Hartland SN, Vernon M, Duffield JS, Benyon RC, Forbes SJ, Iredale JP. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology 2012;55:1965-75.
37. Gonzalez S, Moran M, Kochevar IE. Chronic photodamage in skin of mast cell-deficient mice. Photochem Photobiol 1999;70:248-53.
38. Grimbaldeston MA, Simpson A, Finlay-Jones JJ, Hart PH. The effect of ultraviolet radiation exposure on the prevalence of mast cells in human skin. Br J Dermatol 2003;148:300-6.
39. Farrell DJ, Hines JE, Walls AF, Kelly PJ, Bennett MK, Burt AD. Intrahepatic mast cells in chronic liver diseases. Hepatology 1995;22:1175-81.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Wu J, Lawrence C, Wadsworth RM, Kennedy S. Perivascular mast cells promote neointimal elastin deposition and suppress chronic vein graft restenosis in hyperlipidaemic mice. Vessel Plus 2017;1:137-44. http://dx.doi.org/10.20517/2574-1209.2017.15
AMA Style
Wu J, Lawrence C, Wadsworth RM, Kennedy S. Perivascular mast cells promote neointimal elastin deposition and suppress chronic vein graft restenosis in hyperlipidaemic mice. Vessel Plus. 2017; 1: 137-44. http://dx.doi.org/10.20517/2574-1209.2017.15
Chicago/Turabian Style
Wu, Junxi, Catherine Lawrence, Roger M. Wadsworth, Simon Kennedy. 2017. "Perivascular mast cells promote neointimal elastin deposition and suppress chronic vein graft restenosis in hyperlipidaemic mice" Vessel Plus. 1: 137-44. http://dx.doi.org/10.20517/2574-1209.2017.15
ACS Style
Wu, J.; Lawrence C.; Wadsworth RM.; Kennedy S. Perivascular mast cells promote neointimal elastin deposition and suppress chronic vein graft restenosis in hyperlipidaemic mice. Vessel Plus. 2017, 1, 137-44. http://dx.doi.org/10.20517/2574-1209.2017.15
About This Article
Copyright

Author Biographies

Data & Comments
Data
0

 Cite This Article 0 clicks
Cite This Article 0 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.